×
Close
Sign Up
Login
Home
Library S
Library D
Events & Partner
WeMed
MDLA Events Platform
Events
Media Partners
Educational Partner
User Tools
FAQ/USER GUIDE
Language
English
中文/ Chinese
French
Português
Español
Arabic
Russian
Contact Us
×
Close
mdla_1
mdla_2
mdla_3
mdla_4
mdla_5
mdla_6
Categories
Biotechnology & Applied Microbiology
8589
Global Medical University
5154
Allergy
1797
Anatomy & Morphology
1584
Andrology
379
Anesthesia & Intensive Care
1291
Anesthesiology
5450
Audiology & Speech-Language Pathology
347
Behavioral Sciences
100
Biochemical Research Methods
7105
Biochemistry & Molecular Biology
29887
Biodiversity Conservation
345
Biology
8422
Biophysics
8197
Cardiac & Cardiovascular Systems
30959
Cardiovascular & Respiratory Systems
1374
Cell & Tissue Engineering
702
Cell Biology
11161
Chemistry, Analytical
4284
Chemistry, Applied
10876
Chemistry, Medicinal
8727
Chemistry, Multidisciplinary
18481
Clinical Immunology & Infectious Disease
436
Clinical Medicine
8939
Clinical Neurology
16556
Clinical Psychology & Psychiatry
1302
Critical Care Medicine
3180
Dentistry, Oral Surgery & Medicine
13451
Dermatology
7462
Developmental Biology
6888
Ecology
643
Education, Scientific Disciplines
2062
Emergency Medicine
4069
Endocrinology, Metabolism & Nutrition
24392
Engineering, Biomedical
3738
Entomology
449
Environmental Medicine & Public Health
4703
Evolutionary Biology
267
Gastroenterology & Hepatology
12174
General & Internal Medicine
6920
Genetics & Heredity
15020
Geriatrics & Gerontology
5110
Gerontology
366
Health Care Sciences & Services
16140
Health Policy & Services
647
Hematology
5590
Immunology
24610
Infectious Diseases
13945
Integrative & Complementary Medicine
2938
Medical Ethics
1230
Medical Informatics
2257
Medical Laboratory Technology
419
Medicine, General & Internal
44152
Medicine, Legal
532
Medicine, Research & Experimental
17667
Microbiology
23203
Mycology
0
Nanoscience & Nanotechnology
5449
Neuroimaging
1404
Neurology
4561
Neurosciences
39529
Nursing
9712
Nutrition & Dietetics
7839
Obstetrics & Gynecology
8246
Oncology
52017
Ophthalmology
9693
Optics
4263
Orthopedics
11735
Orthopedics, Rehabilitation & Sports Medicine
1797
Otolaryngology
1589
Otorhinolaryngology
4849
Parasitology
1134
Pathology
5117
Pediatrics
21544
Peripheral Vascular Disease
4802
Pharmacology & Pharmacy
35219
Pharmacology/Toxicology
12055
Physiology
8847
Polymer Science
531
Primary Health Care
870
Psychiatry
19011
Psychology
5213
Psychology, Applied
112
Psychology, Biological
388
Psychology, Clinical
807
Psychology, Developmental
243
Psychology, Educational
169
Psychology, Experimental
166
Psychology, Mathematical
0
Psychology, Multidisciplinary
1682
Psychology, Psychoanalysis
30
Psychology, Social
119
Public Health & Health Care Science
2245
Public, Environmental & Occupational Health
27039
Quantum Science & Technology
0
Radiology, Nuclear Medicine & Imaging
12338
Radiology, Nuclear Medicine & Medical Imaging
8061
Rehabilitation
3031
Remote Sensing
0
Reproductive Biology
2859
Reproductive Medicine
1192
Research/Laboratory Medicine & Medical Technology
3976
Respiratory System
7402
Rheumatology
6032
Social Sciences, Biomedical
1191
Substance Abuse
2741
Surgery
33651
Toxicology
4439
Transplantation
948
Tropical Medicine
300
Urology & Nephrology
12941
Veterinary Sciences
35
Virology
2499
Zoology
0
Channels
GENOME BIOLOGY
666
APPLIED MICROBIOLOGY AND BIOTECHNOLOGY
1239
BIOTECHNOLOGY ADVANCES
467
CURRENT OPINION IN MICROBIOLOGY
325
ENZYME AND MICROBIAL TECHNOLOGY
438
GENOMICS
674
JOURNAL OF BIOTECHNOLOGY
460
NATURE BIOTECHNOLOGY
835
AMB EXPRESS
400
BIOTECHNOLOGY AND APPLIED BIOCHEMISTRY
215
CYTOTECHNOLOGY
218
ELECTRONIC JOURNAL OF BIOTECHNOLOGY
168
FOLIA MICROBIOLOGICA
299
GENOME
102
INTERNATIONAL JOURNAL OF BIOLOGICAL MARKERS
26
IRANIAN JOURNAL OF BIOTECHNOLOGY
115
JOURNAL OF APPLIED MICROBIOLOGY
414
JOURNAL OF BIOLOGICAL ENGINEERING
139
JOURNAL OF FOOD SAFETY
51
JOURNAL OF GENE MEDICINE
57
JOURNAL OF GENERAL VIROLOGY
405
LETTERS IN APPLIED MICROBIOLOGY
157
JOURNAL OF VIROLOGICAL METHODS
517
BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS
202
SCI Abstract
search
ALL
RECOMMENDED
+
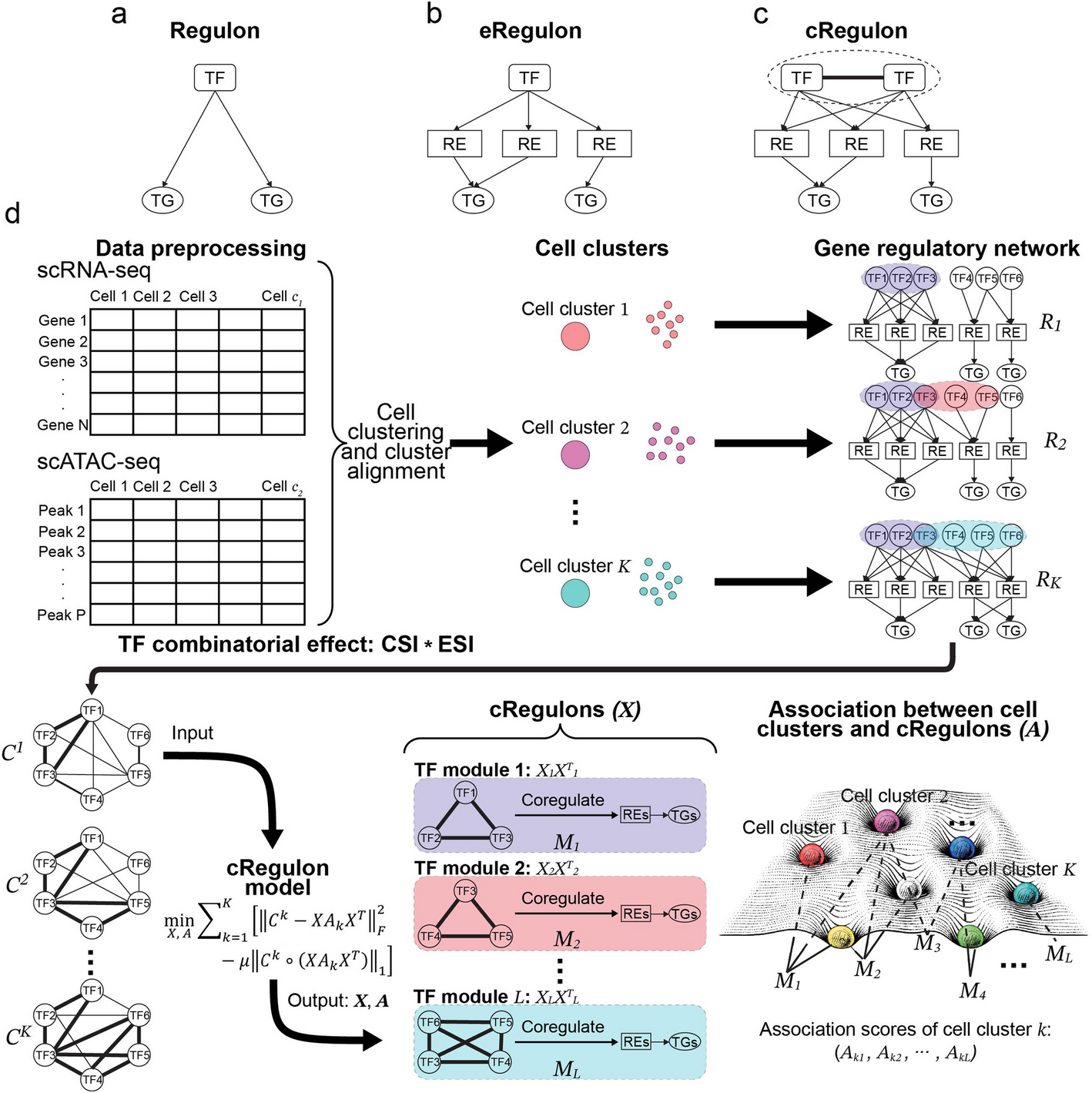
Modeling combinatorial regulation from single-cell multi-omics provides regulatory units underpinning cell type landscape using cRegulon
Advances in single-cell technology enable large-scale generation of omics data, promising for clarifying gene regulatory n...
Genome Biology
comment
0
thumb_up
0
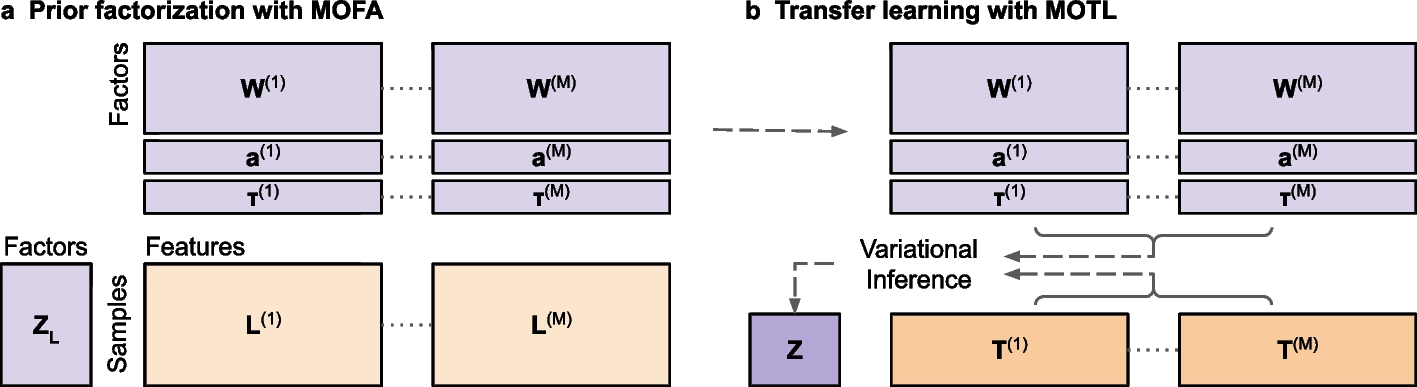
MOTL: enhancing multi-omics matrix factorization with transfer learning
Joint matrix factorization is popular for extracting lower dimensional representations of multi-omics data but loses effec...
Genome Biology
comment
0
thumb_up
0
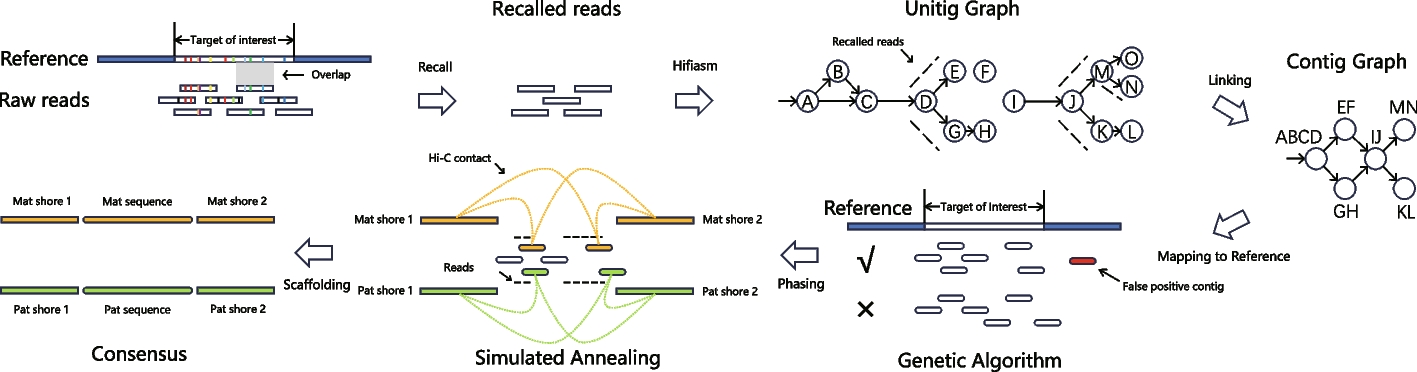
TRFill: synergistic use of HiFi and Hi-C sequencing enables accurate assembly of tandem repeats for population-level analysis
The highly repetitive content of eukaryotic genomes, including long tandem repeats, segmental duplications, and centromere...
Genome Biology
comment
0
thumb_up
0
Field-crop transcriptome models are enhanced by measurements in systematically controlled environments
Plants in the field respond to seasonal and diel changes in various environmental factors such as irradiance and temperatu...
Genome Biology
comment
0
thumb_up
0
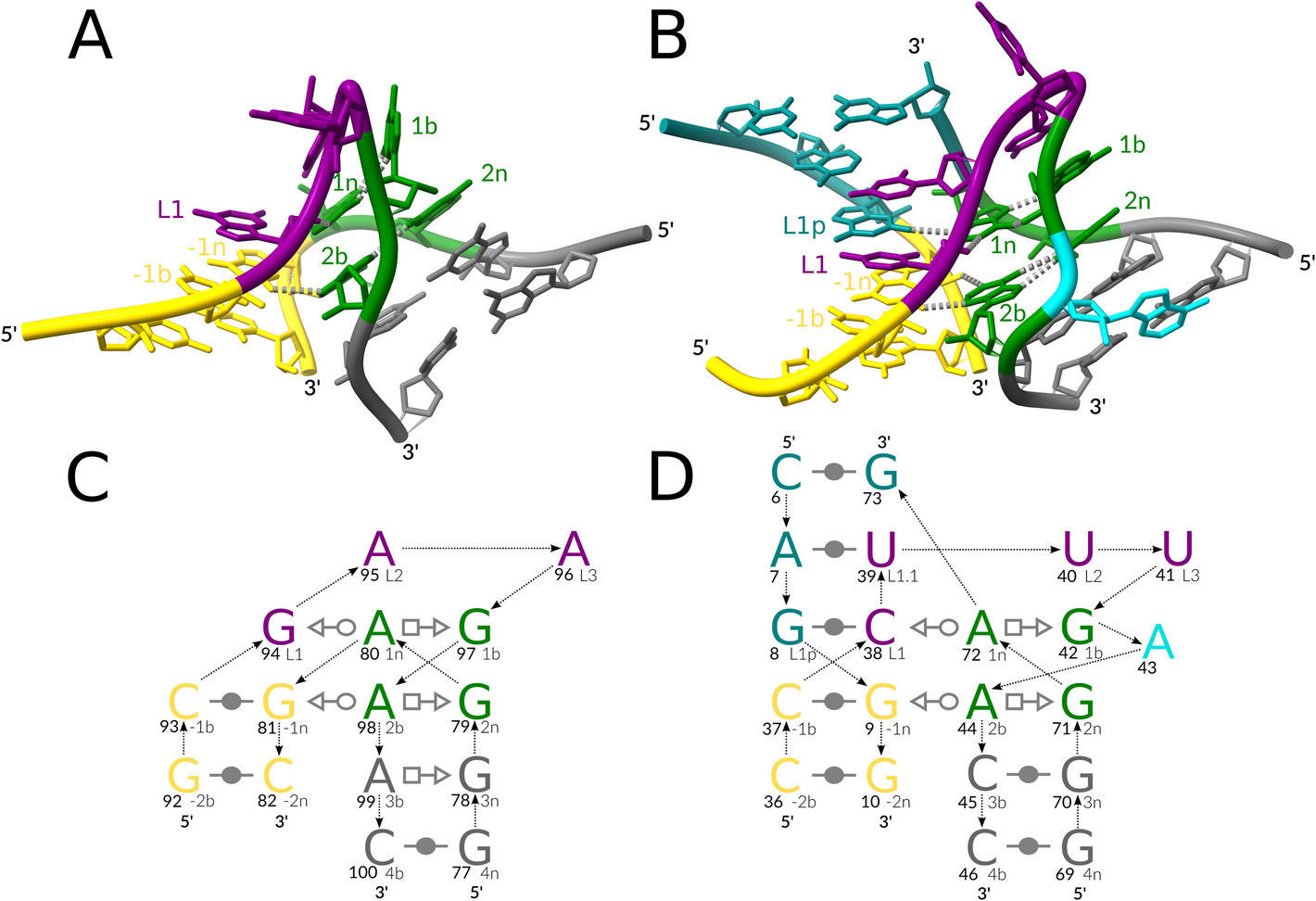
ARTEM: a method for RNA and DNA tertiary motif identification with backbone permutations
Non-coding RNA functions are largely defined by their 3D structures, which consist of recurrent building blocks, tertiary ...
Genome Biology
comment
0
thumb_up
0
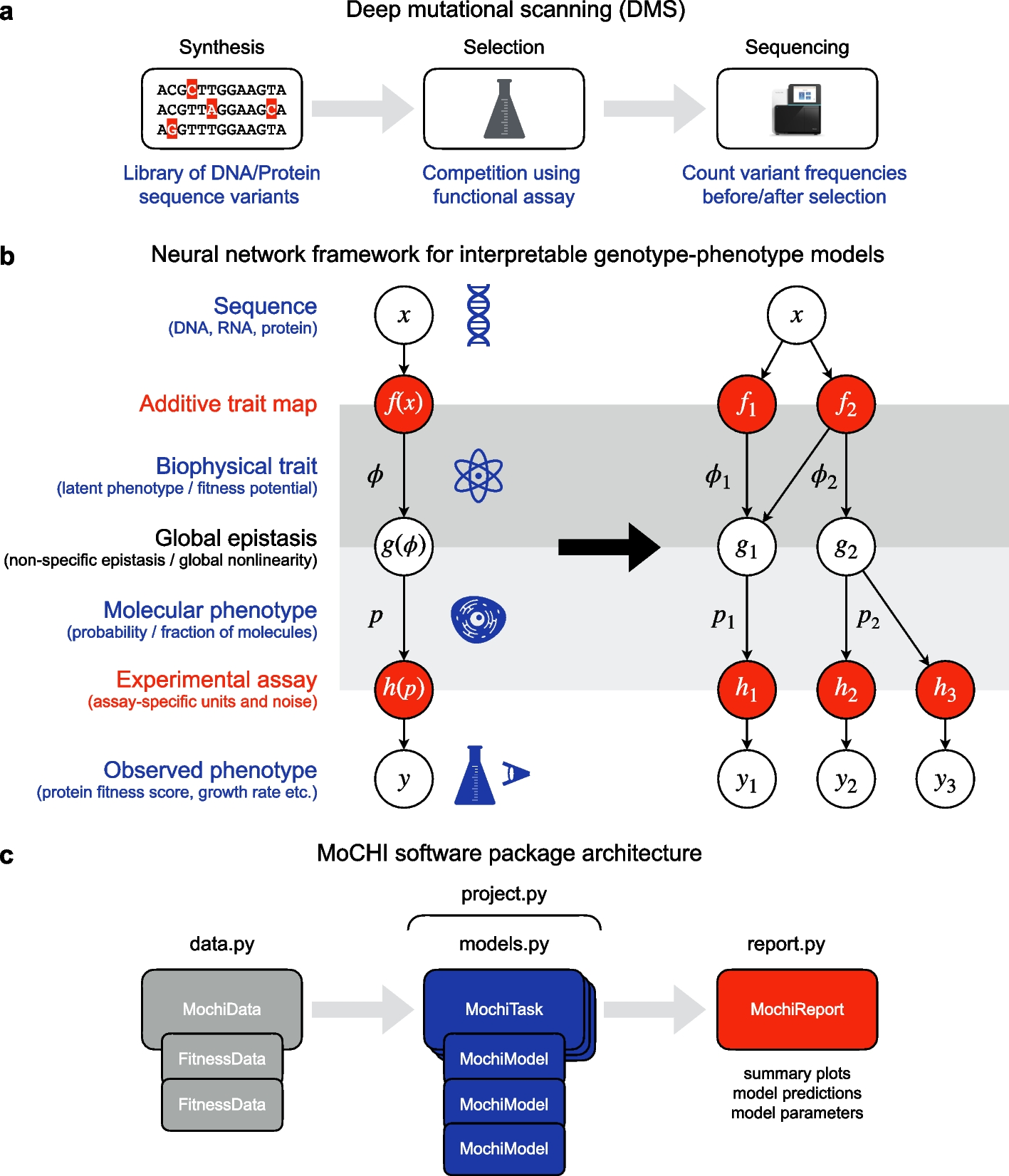
MoCHI: neural networks to fit interpretable models and quantify energies, energetic couplings, epistasis, and allostery from deep mutational scanning data
We present MoCHI, a tool to fit interpretable models using deep mutational scanning data. MoCHI infers free energy changes...
Genome Biology
comment
0
thumb_up
0
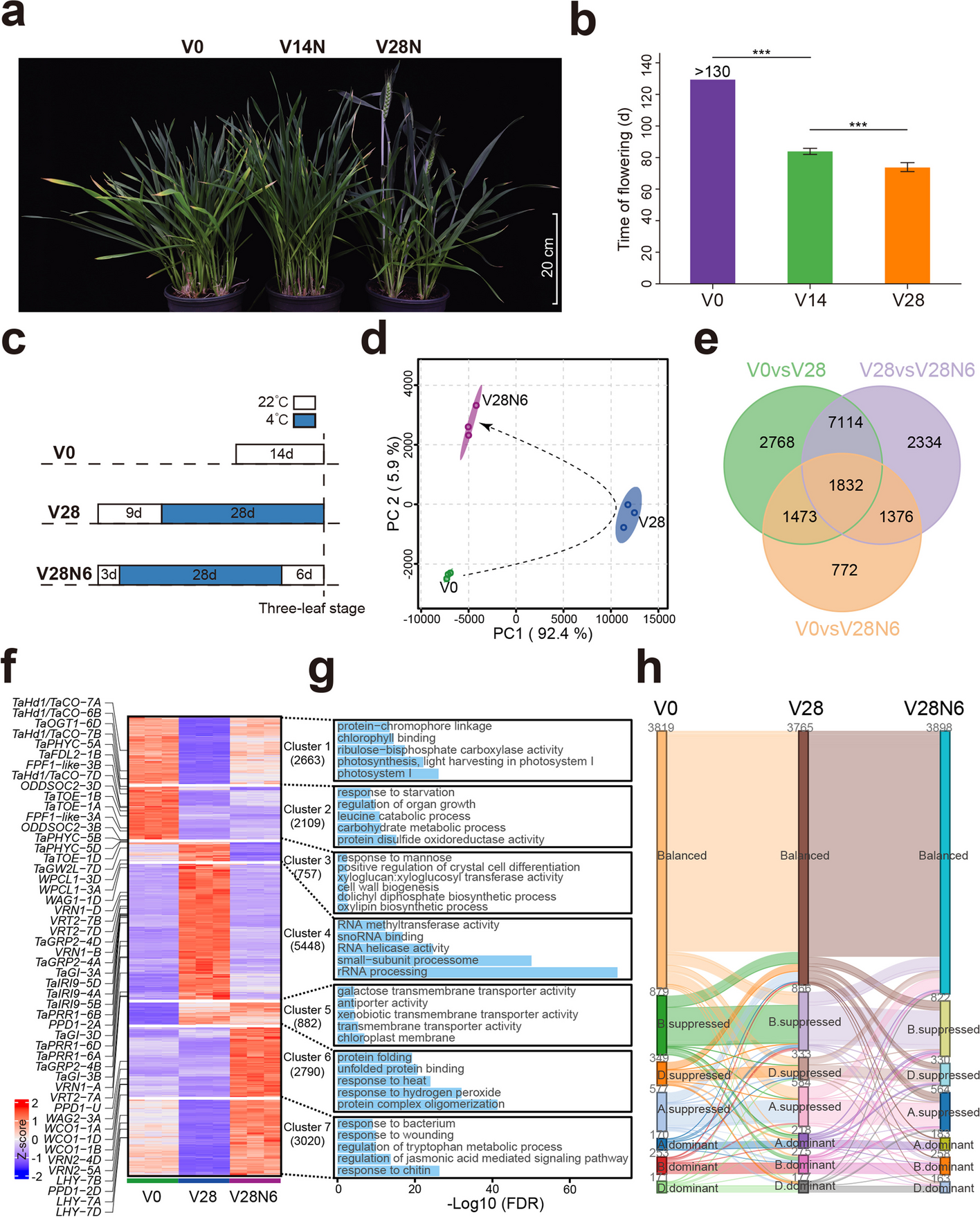
Chromatin loops gather targets of upstream regulators together for efficient gene transcription regulation during vernalization in wheat
Plants respond to environmental stimuli by altering gene transcription that is highly related with chromatin status, inclu...
Genome Biology
comment
0
thumb_up
0
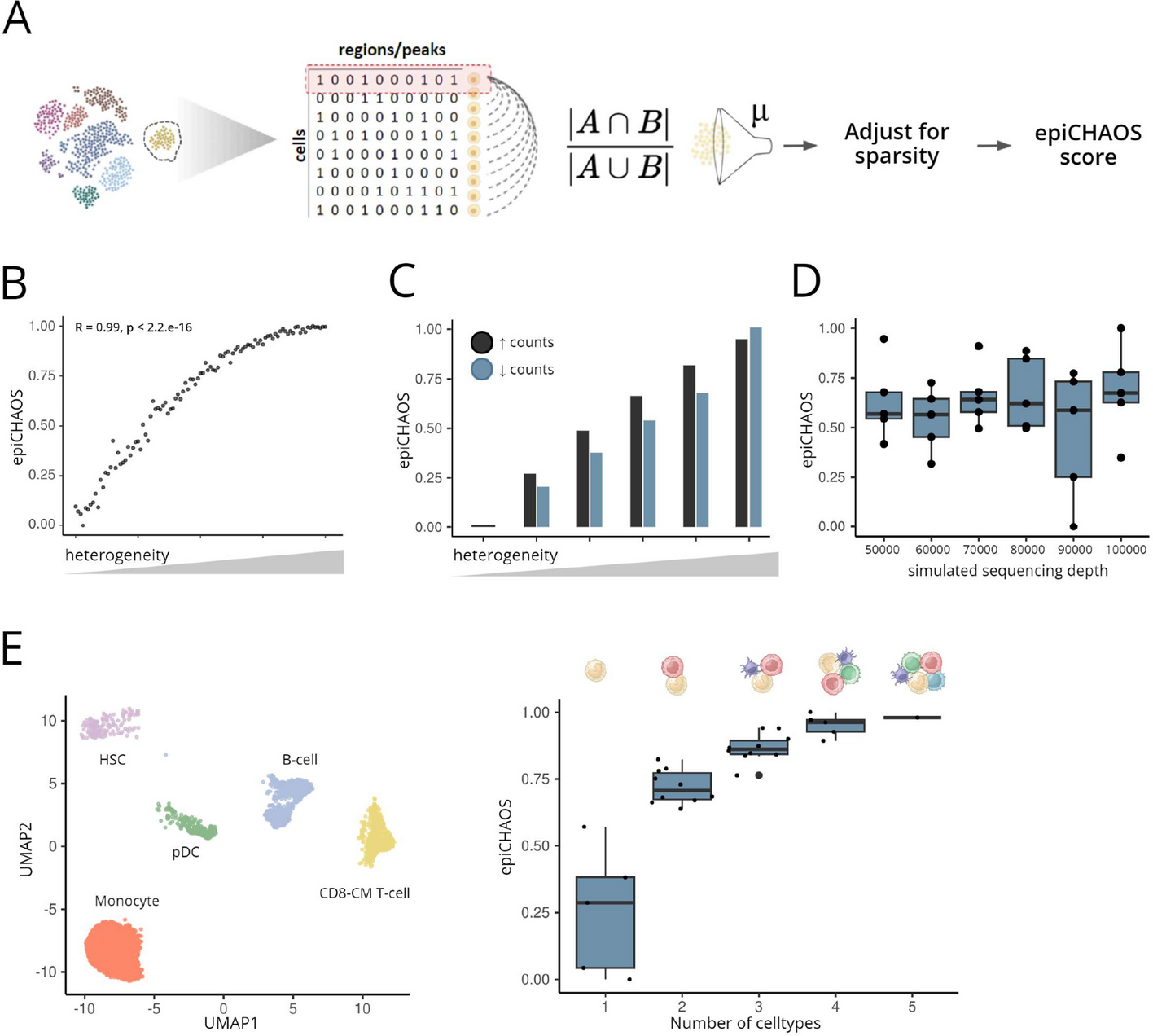
EpiCHAOS: a metric to quantify epigenomic heterogeneity in single-cell data
Epigenetic heterogeneity is a fundamental property of biological systems and is recognized as a potential driver of tumor ...
Genome Biology
comment
0
thumb_up
0
Increased spatial coupling of integrin and collagen IV in the immunoresistant clear-cell renal-cell carcinoma tumor microenvironment
Immunotherapy has improved survival for patients with advanced clear cell renal cell carcinoma (ccRCC), but resistance to ...
Genome Biology
comment
0
thumb_up
0
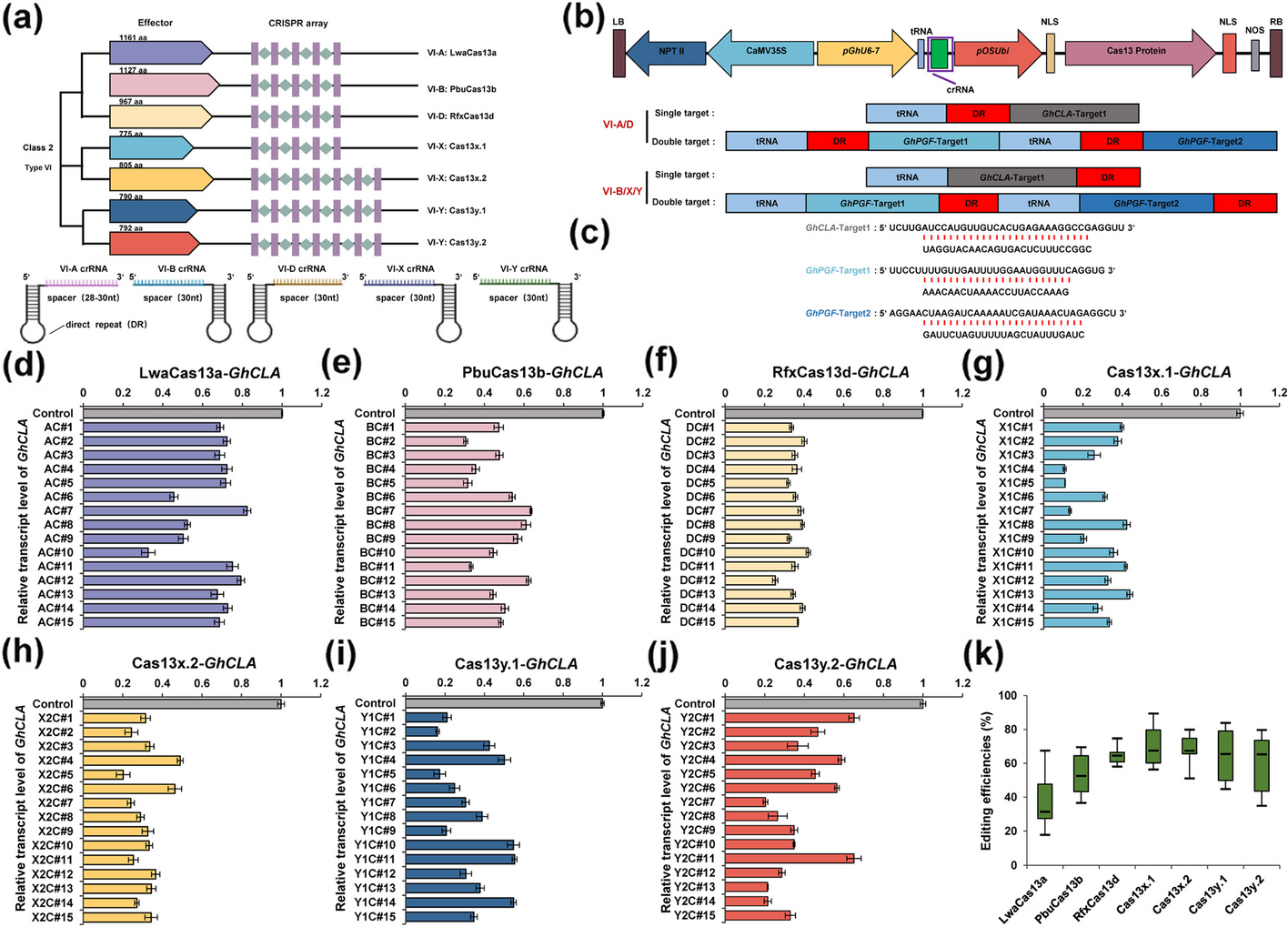
Systemic evaluation of various CRISPR/Cas13 orthologs for knockdown of targeted transcripts in plants
CRISPR/Cas13 system, recognized for its compact size and specificity in targeting RNA, is currently employed for RNA degra...
Genome Biology
comment
0
thumb_up
0
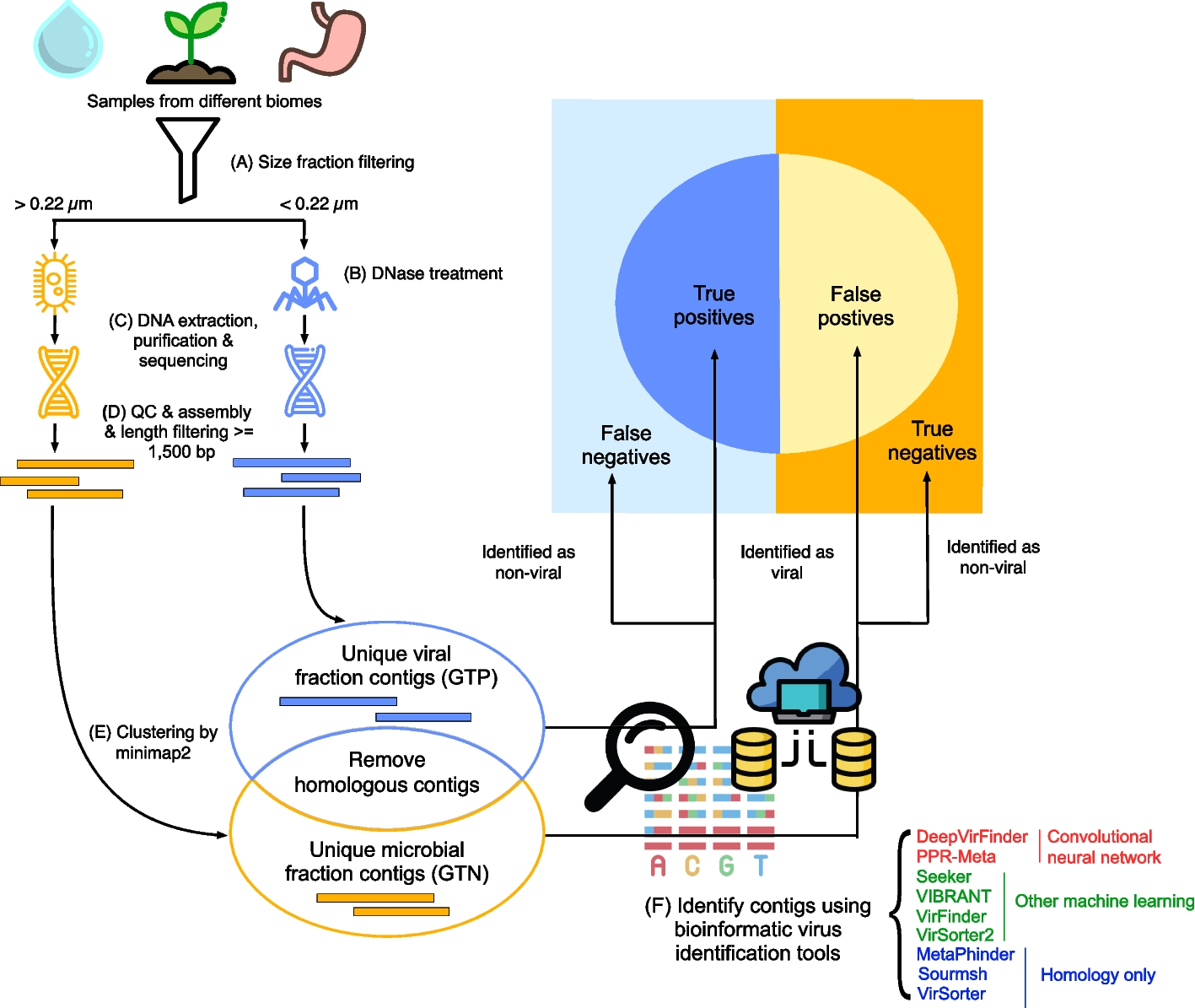
Benchmarking bioinformatic virus identification tools using real-world metagenomic data across biomes
As most viruses remain uncultivated, metagenomics is currently the main method for virus discovery. Detecting viruses in m...
Genome Biology
comment
0
thumb_up
0
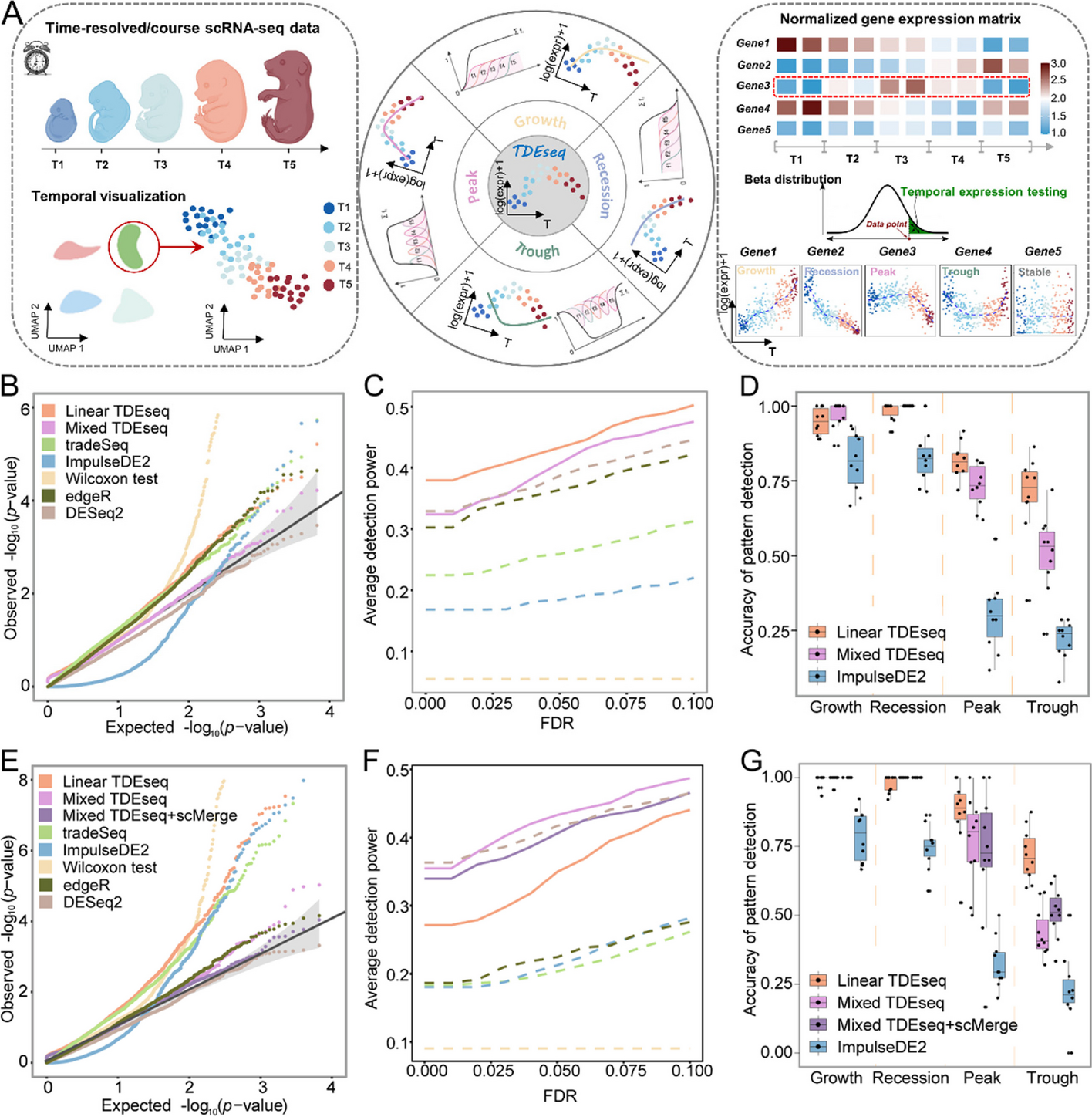
Powerful and accurate detection of temporal gene expression patterns from multi-sample multi-stage single-cell transcriptomics data with TDEseq
We present a non-parametric statistical method called TDEseq that takes full advantage of smoothing splines basis function...
Genome Biology
comment
0
thumb_up
0
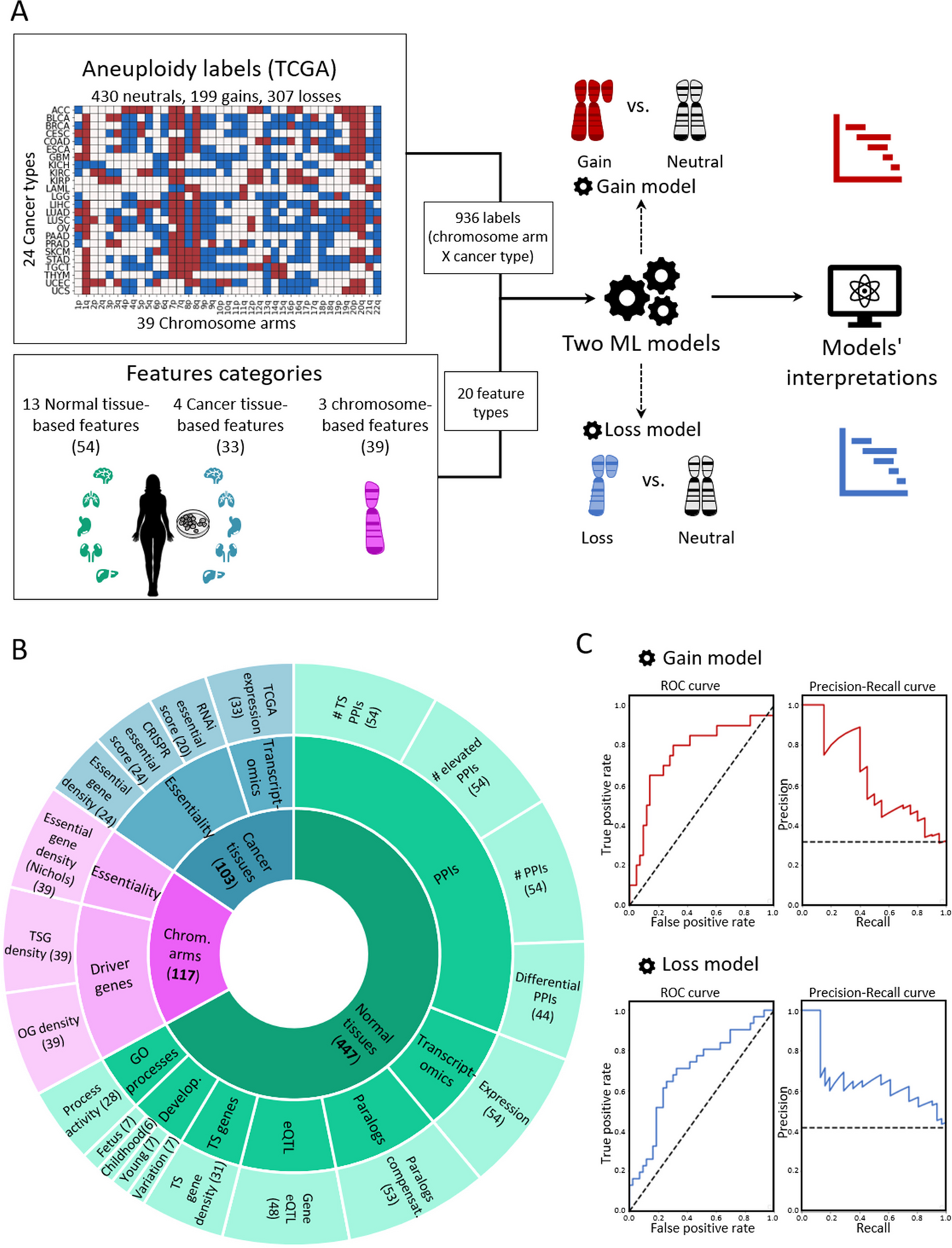
Machine-learning analysis reveals an important role for negative selection in shaping cancer aneuploidy landscapes
Aneuploidy, an abnormal number of chromosomes within a cell, is a hallmark of cancer. Patterns of aneuploidy differ across...
Genome Biology
comment
0
thumb_up
0
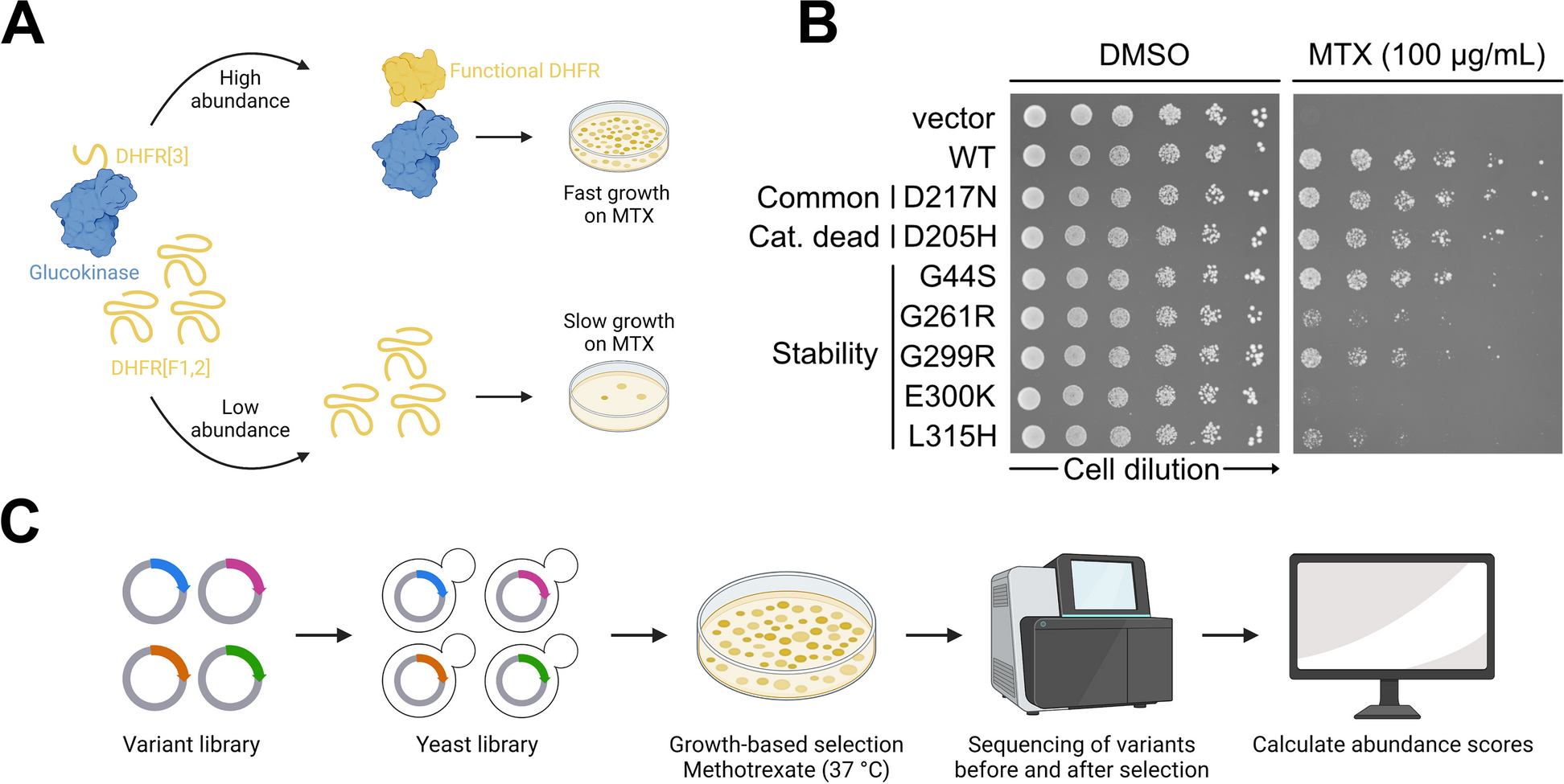
Characterizing glucokinase variant mechanisms using a multiplexed abundance assay
Amino acid substitutions can perturb protein activity in multiple ways. Understanding their mechanistic basis may pinpoint...
Genome Biology
comment
0
thumb_up
0
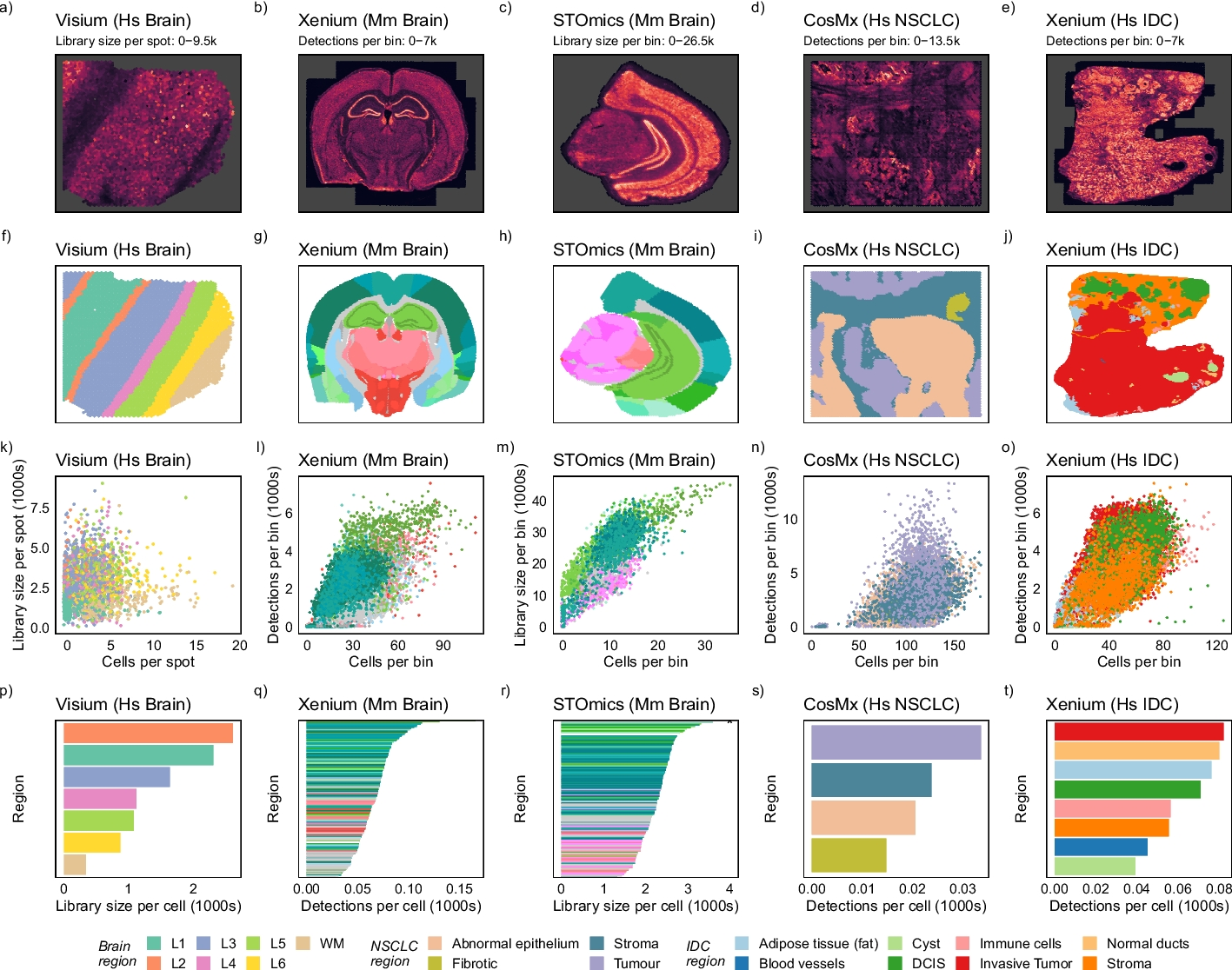
Library size confounds biology in spatial transcriptomics data
Spatial molecular data has transformed the study of disease microenvironments, though, larger datasets pose an analytics c...
Genome Biology
comment
0
thumb_up
0
SQANTI-SIM: a simulator of controlled transcript novelty for lrRNA-seq benchmark
Long-read RNA sequencing has emerged as a powerful tool for transcript discovery, even in well-annotated organisms. Howeve...
Genome Biology
comment
0
thumb_up
0
DNA methylation modulated genetic variant effect on gene transcriptional regulation
Expression quantitative trait locus (eQTL) analysis has emerged as an important tool in elucidating the link between genet...
Genome Biology
comment
0
thumb_up
0
Turnover of strain-level diversity modulates functional traits in the honeybee gut microbiome between nurses and foragers
Strain-level diversity is widespread among bacterial species and can expand the functional potential of natural microbial ...
Genome Biology
comment
0
thumb_up
0
Pediatric glioma histone H3.3 K27M/G34R mutations drive abnormalities in PML nuclear bodies
Point mutations in histone variant H3.3 (H3.3K27M, H3.3G34R) and the H3.3-specific ATRX/DAXX chaperone complex are frequen...
Genome Biology
comment
0
thumb_up
0
Cotton pedigree genome reveals restriction of cultivar-driven strategy in cotton breeding
Many elite genes have been identified from the available cotton genomic data, providing various genetic resources for gene...
Genome Biology
comment
0
thumb_up
0
GCLiPP: global crosslinking and protein purification method for constructing high-resolution occupancy maps for RNA binding proteins
GCLiPP is a global RNA interactome capture method that detects RNA-binding protein (RBP) occupancy transcriptome-wide. GCL...
Genome Biology
comment
0
thumb_up
0
Meiosis in an asymmetric dikaryotic genome of Tremella fuciformis Tr01 facilitates new chromosome formation
The dikaryotic stage dominates most of the life cycle in basidiomycetes, and each cell carries two different haploid nucle...
Genome Biology
comment
0
thumb_up
0
Dengue and Zika RNA-RNA interactomes reveal pro- and anti-viral RNA in human cells
Identifying host factors is key to understanding RNA virus pathogenicity. Besides proteins, RNAs can interact with virus g...
Genome Biology
comment
0
thumb_up
0
Integration of datasets for individual prediction of DNA methylation-based biomarkers
Epigenetic scores (EpiScores) can provide biomarkers of lifestyle and disease risk. Projecting new datasets onto a referen...
Genome Biology
comment
0
thumb_up
0
A large-scale genomically predicted protein mass database enables rapid and broad-spectrum identification of bacterial and archaeal isolates by mass spectrometry
MALDI-TOF MS-based microbial identification relies on reference spectral libraries, which limits the screening of diverse ...
Genome Biology
comment
0
thumb_up
0
Reconstruction of private genomes through reference-based genotype imputation
Genotype imputation is an essential step in genetic studies to improve data quality and statistical power. Public imputati...
Genome Biology
comment
0
thumb_up
0
Haplotype-resolved assemblies and variant benchmark of a Chinese Quartet
Recent state-of-the-art sequencing technologies enable the investigation of challenging regions in the human genome and ex...
Genome Biology
comment
0
thumb_up
0
TargetRNA3: predicting prokaryotic RNA regulatory targets with machine learning
Small regulatory RNAs pervade prokaryotes, with the best-studied family of these non-coding genes corresponding to trans-a...
Genome Biology
comment
0
thumb_up
0
Hybrid-hybrid correction of errors in long reads with HERO
Although generally superior, hybrid approaches for correcting errors in third-generation sequencing (TGS) reads, using nex...
Genome Biology
comment
0
thumb_up
0
Comparing methods for constructing and representing human pangenome graphs
As a single reference genome cannot possibly represent all the variation present across human individuals, pangenome graph...
Genome Biology
comment
0
thumb_up
0
Load More
Modal title
×
Modal title
×
Share
Login
Global News and Health Forum
Join Now!
Member Login
Remember me
Forgot password?
Or using
Linkedin