
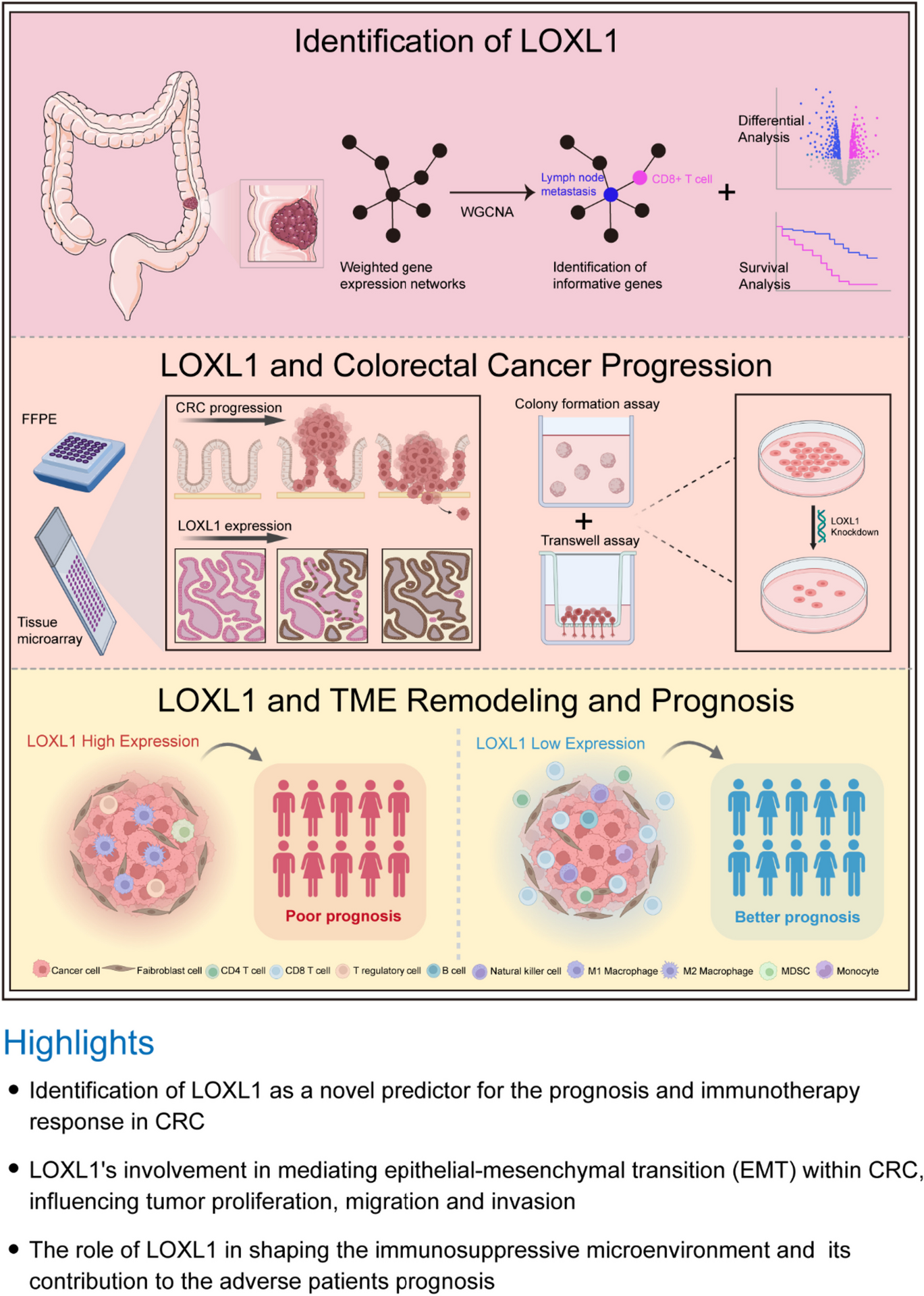
記住我
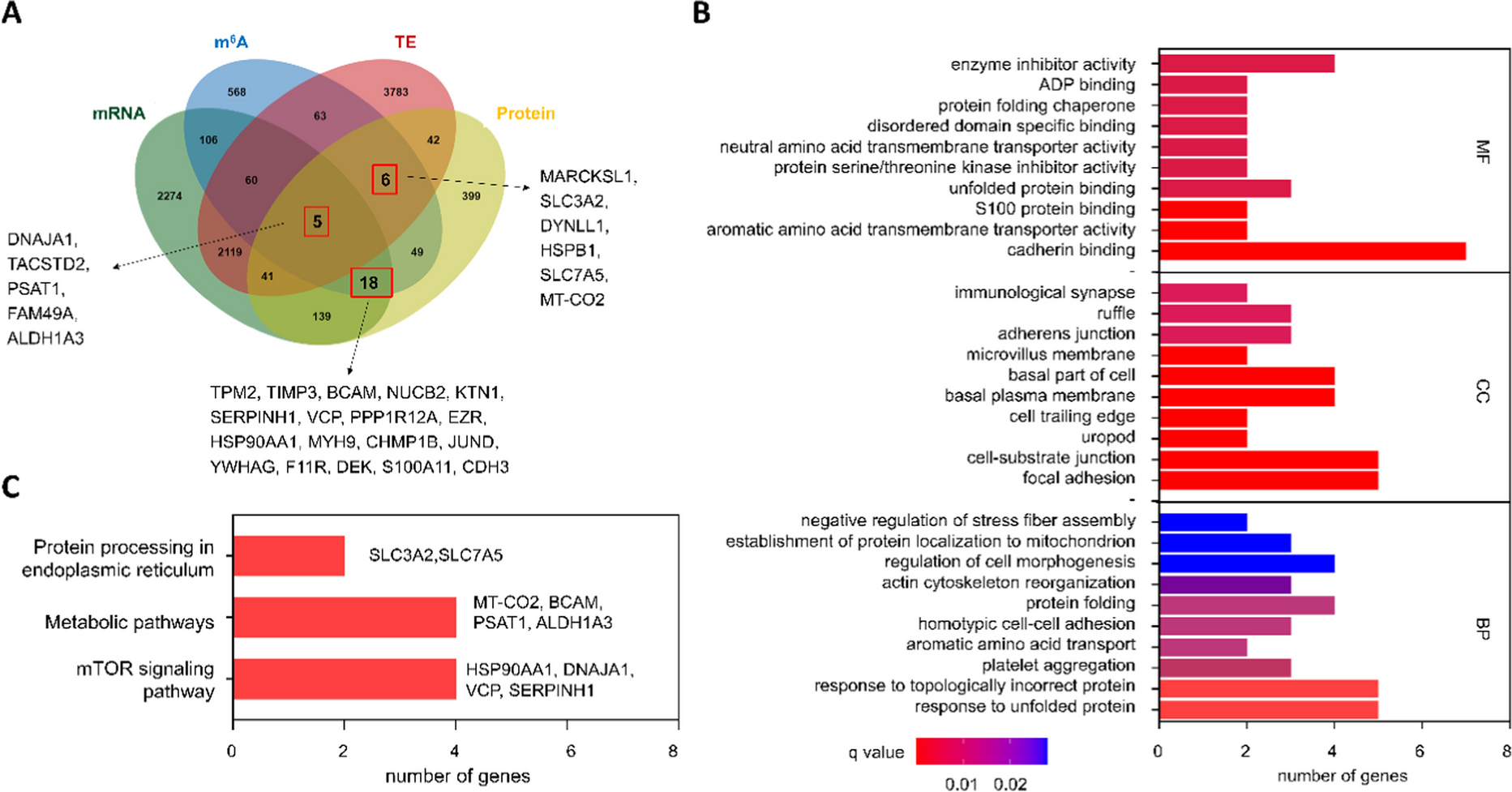
Since the CD8 + T cell infiltration plays an indispensable role in CRC prognosis, we integrated CD8 + T cell levels (assessed via CIBERSORT) with clinical traits to perform WGCNA. We included a total of 19,028 gene expression profiles of 237 samples from GSE39582 discovery dataset to construct co-expression network after removing outlier samples (Supplementary Fig. 1A). For constructing a scale-free network, we chose a power of β = 4 (yielding a scale-free R2 = 0.9) (Supplementary Fig. 1B). WGCNA clustered all these genes into 12 gene modules using a cluster dendrograms (Supplementary Fig. 1C). Notably, the turquoise module exhibited the highest correlation with both CD8 + T cell infiltration and pN stage in CRC (R = -0.21, P = 4e-04; R = 0.19, P = 0.001, respectively) (Fig. 1A), suggesting genes in the turquoise module may be closely correlated with the malignancy and prognosis of CRC. Scatter plots of MM vs. GS verified the association between turquoise module and CD8 + T cells infiltration or pN stage (Supplementary Fig. 1D-E), leading to its identification as a key module. Subsequent GO and KEGG analyses revealed that the turquoise module genes predominantly participate in wound healing, extracellular matrix organization, epithelial to mesenchymal transition (Fig. 1B, Table S4), and pathways including focal adhesion, TGF-beta signaling, and ECM-receptor interaction, etc. (Fig. 1C, Table S4), indicating the hub module genes might serve critical functions in mediating the TME and metastasis of CRC.
Fig. 1
Screening of hub module via weighted gene co-expression network analysis (WGCNA) and identification of LOXL1 as a hub gene. (A) Left: Heatmap for the correlation between module eigengenes and clinical traits including pathological T, N stage and CD8 + T cell infiltration level of colorectal cancer (CRC) patients in GSE39582 discovery dataset. Each cell contains corresponding correlation coefficient and P-value. P-values were calculated using Pearson’s correlation analysis. The turquoise module was selected as the most significant module which was positively correlated with pN stage and negatively correlated with CD8 + T cell infiltration. Right: The bar chart indicates the number of genes in each module. (B) Gene Ontology (GO) biological process (BP) enrichment analysis and (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for genes in the turquoise module. A p-value less than 0.05 indicated statistical significance. (D) The GSE39582 validation dataset which contains 19 colorectal adjacent normal tissues and 123 colorectal tumor tissues was used to examine the differentially expressed genes (DEGs) between normal and tumor tissues. 814 DEGs including 469 up-regulated and 345 down-regulated genes were identified and selected to draw a volcano plot. The red and blue dots represent significantly up-regulated and down-regulated genes respectively (|log2FC|> 1.5, P < 0.01), and grey dots represent genes without significant expression changes. (E) The Venn diagram depicted the overlapping genes between up-regulated DEGs and genes in the turquoise module. A total of 23 overlapping hub genes were obtained. (F) The circle plot determined that LOXL1 was the only gene significantly associated with patient’s overall survival in the GSE17536 cohort, GSE39582 cohort and TCGA-COAD cohort. (G) The Kaplan–Meier curves showed that high expression of LOXL1 was correlated with poor survival rate of CRC patients in GSE17536 cohort, GSE39582 cohort, and TCGA-COAD cohort. Optimal separation cut-off value was used to achieve best statistical significance. (H) Univariate and multivariate Cox proportional hazards regression analysis showed independent factors for overall survival (OS) in TCGA-COAD cohort, GSE39582 cohort and GSE17536 cohort. Forest plot presents the hazard ratio (HR) value and 95% confidence interval (CI). (I) A nomogram combining LOXL1 expression and pathological stage was constructed to predict the 1-, 3-, and 5-year overall survival probability of CRC patients. The red line and arrows represent an example of designated points. (J) Calibration curves were used to validate the consistency between predicted nomogram results and the actual 1-, 3-, and 5-year survival outcomes. The y-axis represents the measured survival probabilities. The x-axis represents the nomogram-predicted survival probabilities. The diagonal grey solid line represents the ideal nomogram, and the blue, green, purple line represents the 1-, 3-, and 5-year observed nomograms respectively
Identification of LOXL1 as a prognostic hub gene in CRCTo determine the hub genes responsible for CRC prognosis, we firstly examined the GSE39582 cohort and identified 814 DEGs between tumor and para-tumor tissues (Fig. 1D, Table S5), which were found to be involved in mediating signaling like extracellular matrix organization and ECM-receptor interaction (Supplementary Fig. 2A and B, Table S6). Intersection of the 469 up-regulated DEGs with 134 genes from the turquoise module (selected based on MM > 0.8 and GS > 0.2 criteria) led to the identification of 23 candidate genes (Fig. 1E). To assess the prognostic relevance of these 23 genes, KM survival analysis on patients stratified by the expression levels of these 23 genes was implemented in three independent cohorts. The result showed that LOXL1 was the only gene that might affect clinical outcome (Fig. 1F, Table S7). The KM curve in the three cohorts demonstrated that high LOXL1 expression was significantly linked with r worse OS (Fig. 1G). To further confirm the prognosis prediction performance of LOXL1, its association with OS in CRC patients was evaluated using univariate and multivariate Cox regression analysis. The analyses confirmed that high LOXL1 expression, along with advanced pathological stage, were significant risk factors associated with poor prognosis. Notably, LOXL1 expression remained an independent prognostic factor even after adjustment for other clinical risk factors (Fig. 1H). Additionally, with a goal of predicting the survival probability for CRC patients in clinic, a risk estimation nomogram based on the above two independent risk factors (tumor stage and LOXL1) was established and scores were calculated to predict 1-, 3- and 5-year OS for individual patient (Fig. 1I). Calibration plots revealed a high level of concordance between predictive and observed outcomes, indicating the good performance of the nomogram in predicting patient’s OS (Fig. 1J).
LOXL1 expression was positively correlated with TNM staging and poor differentiationThe role of LOXL1 in CRC malignancy was evaluated. In TCGA-COAD cohort, the distribution of clinicopathological features including age, gender, stage, histological type, survival status, OS time, neoadjuvant treatment, and radiation therapy were examined in patient groups with high or low LOXL1 expression. The results suggested that elevated LOXL1 expression was inversely associated with OS duration (Fig. 2A, Table S8), and markedly higher in patients exhibiting advanced pT and pN stage, while no significant difference was observed for the pM stage (Fig. 2B-D). Similar results were obtained when analyzing the GSE39582 cohort. Larger tumor size (T3-4), regional lymph nodes metastasis (N2-3) and advanced tumor stage (stage III-IV) were more commonly detected in patients with high LOXL1 expression (Fig. 2E). Furthermore, quantitative RT-PCR analysis of 38 clinical sample pairs revealed a pronounced upregulation of LOXL1 mRNA in CRC tissues, compared with non-tumor tissues (Fig. 2F). IHC assessments on a tissue microarray containing 208 pairs of CRC tissues corroborated the augmented LOXL1 protein expression in tumor tissues, with a significant association to poorer differentiation status (Fig. 2G and H).
Fig. 2
The correlation between LOXL1 expression and clinicopathological characteristics of CRC patients. (A) Heatmap visualizing the distribution of clinicopathological features in patients divided by the expression level of LOXL1 from TCGA-COAD cohort. White lines represent missing values. (B-D) The LOXL1 expression level in TCGA CRC patients stratified by pathological T, N, and M stage. (E) The circular pie chart shows the proportion difference of clinical indices between LOXL1 high and low expression groups from the GSE39582 cohort. Chi-squared test was used for statistical analysis, ns: not significant, *P < 0.05, **P < 0.01, ***P < .001. (F) Relative mRNA expression of LOXL1 was quantified in a cohort of surgically resected human CRC tissues and paired non-tumor tissues (n = 38) through quantitative real-time PCR, **P < 0.01. Statistical significance was determined by the paired student’s t test and error bars represent standard deviations. (G) IHC staining for LOXL1 expression was performed on tissue microarray (TMA) containing normal colon tissues, well differentiated- and poorly differentiated CRC tissues from the SYSU cohort. (H) (left) Quantification of LOXL1 expression in normal or tumor tissues based on IHC results of the TMA. H-score represents the immunostaining score obtained by Image Pro Plus software. Statistical significance was assessed using the unpaired student’s t test. (right) High expression of LOXL1 significantly associated with poor differentiation status (Chi-squared test, *P < 0.05). “Poorly” represents poor and moderate-poor differentiation status, and “Well” represents well and well-moderate differentiation status
LOXL1 was involved in mediating epithelial-mesenchymal transition in CRCTo investigate the association between LOXL1 expression and tumor biological functions, GSVA was performed in two independent cohorts. Results demonstrated that carcinogenic activation-related pathways, such as epithelial-mesenchymal transition (EMT), focal adhesion, ECM receptor interaction, angiogenesis, and TGF-β signaling were predominantly activated in the LOXL1 high expression group, while the NK cell mediated cytotoxicity was overrepresented in LOXL1 low group (Fig. 3A and B), indicating the involvement of LOXL1 in mediating immune cell function and tumorigenesis.
Fig. 3
Biological pathway enrichment analysis of LOXL1 and its correlation with epithelial-mesenchymal transition (EMT) in CRC. (A-B) Gene set variation analysis (GSVA) revealed the activation or inhibition status of HALLMARK terms and KEGG pathways in LOXL1-high and LOXL1-low groups from GSE17536 (A) and GSE161158 (B) cohorts. Samples were categorized according to the median expression level of LOXL1 gene. The statistical significance of differences was determined by Student’s t test. (C) Gene set enrichment analysis (GSEA) revealed the activation or inhibition of signaling pathways in LOXL1-high or LOXL1-low groups from the TCGA-COAD database. (D) Expression levels of EMT associated genes in non-tumor and CRC samples from the SYSU cohort by qRT-PCR, ****P < .0001. The Student’s t test was used for statistical analysis. (E) Heatmap depicting the differential expression pattern of epithelial or mesenchymal markers in LOXL1-high and LOXL1-low groups from the TCGA-COAD cohort. Pearson correlation coefficient of gene expression between LOXL1 and epithelial or mesenchymal markers are shown on the right. (F) Scatter plots demonstrating the positive correlation between LOXL1 and EMT related genes (FN1, TWIST1, VIM, ZEB1, ZEB2) from the SYSU cohort by qRT-PCR. P-values were determined using Pearson correlation analysis
Then we performed GSEA analysis in TCGA-COAD dataset. The top 21 enriched pathways in either LOXL1 high or low expression group were obtained (Table S9). As expected, programs associated with tumor promotion, migration and invasion, as well as proinflammatory responses were prominently enriched in the high LOXL1 expression group, while DNA damage repair associated pathways were upregulated in LOXL1 low expression group (Fig. 3C), suggesting the oncogenic function of LOXL1 in CRC. Given the known involvement of lysyl oxidase (LOX) family members in EMT, the relationship between LOXL1 expression and various EMT markers was examined. A variety of markers were selected based on their relatively high expression in CRC tissues (Fig. 3D). Results demonstrated that LOXL1 correlated positively with various mesenchymal markers and negatively with epithelial markers in TCGA-CRC database (Fig. 3E). Furthermore, qRT-PCR was performed in our own cohort to validate the result. Moderate/strong correlations were observed between LOXL1 and FN1, TWIST1, VIM, ZEB1, and ZEB2 in 38 CRC tissues (Fig. 3F).
LOXL1 expression was significantly associated with TMB and genomic instabilitySince genomic instability and TMB are hallmarks of malignancy (Hanahan and Weinberg 2011), we examined the impact of TMB on CRC prognosis and found no marked difference in OS when comparing high versus low TMB groups (Supplementary Fig. 3A). Intriguingly, elevated LOXL1 expression corresponded with increased TMB, as evidenced by our findings (Supplementary Fig. 3B and C). This link was statistically significant, with a Spearman coefficient R of 0.11 and a P-value of 0.026 (Supplementary Fig. 3D). The distribution of CRC somatic variants in LOXL1-high or low-expression patient groups were profiled. Missense mutations were the most common mutation type (Supplementary Fig. 3E), and C > T occurred most frequently among all single-nucleotide variants (Supplementary Fig. 3F). In addition, an analysis of the top 20 driver genes with the highest mutation frequencies revealed a pattern of co-occurrence (Supplementary Fig. 3G). Not surprisingly, patients with increased LOXL1 expression exhibited a higher overall mutation frequency compared to those with lower expression (Supplementary Fig. 3H and I, Table S10).
LOXL1 promoted CRC proliferation, migration and invasionOur investigation into LOXL1's oncogenic role in CRC involved loss-of-function studies. We initially assessed LOXL1's mRNA and protein levels across various CRC cell lines using qRT-PCR and Western blot analyses (Fig. 4A and B). CRC cell lines with relatively high LOXL1 expression (SW480 and SW620) were selected for loss-of-function studies. The silencing effect of LOXL1 by two shRNAs was determined (Fig. 4C and D). Compared with control cells (shNTC-transfected cells), knockdown of LOXL1 (both shc- and shd- transfected cells) significantly impaired proliferation, colony formation in soft agar, foci formation, migratory and invasive capabilities of CRC cells (Fig. 4E-I). These findings collectively underscore LOXL1's role in promoting CRC proliferation and its metastatic properties.
Fig. 4
Loss of LOXL1 reduces proliferation, migration and invasion of CRC cells in vitro. (A-B) Relative mRNA (A) and protein (B) levels of LOXL1 in various colorectal cancer cell lines were quantified. 18S was used as a loading control for qRT-PCR, and β-tubulin was used as a loading control for Western blot. (C-D) Silencing efficiency of two shRNAs targeting LOXL1 (shc, shd) in SW480 and SW620 cells was assessed using qRT-PCR (C) and western blot (D), with β-actin serving as the loading control for Western blot. (E) The effect of LOXL1 silencing on SW480 and SW620 cells proliferation were evaluated by the CCK-8 assay. Statistical significance: *P < 0.05, **P < 0.01. (F) Representative images of the soft agar colony formation assay in SW480 and SW620 cells after transfection with shNTC or shLOXL1 (left). Quantification analysis of clonogenicity was depicted in the bar chart (right). Statistical significance: *P < 0.05. (G-I) SW480 and SW620 cells transfected with shNTC or shc/shd targeting LOXL1 were subjected to foci colony formation (G), migration (H), and invasion assays (I). Quantification analysis of experimental results are presented in the lower panel. The unpaired students’ t test was used for statistical analysis, ***P < .001
Elevated LOXL1 expression correlate with diminished CD8 + T Cell infiltrationComprehensive annotation of biological processes and signaling pathways suggested the role for LOXL1 in modulating the tumor immune microenvironment in CRC. Utilizing the CIBERSORT algorithm, we explored the relationship between LOXL1 expression and immune cell infiltration. It was observed that adaptive immune cell, notably CD8 + T cells known for their positive impact on survival and immunotherapy response in various cancers (Bruni et al. 2020), was markedly lower in cases with heightened LOXL1 expression (Fig. 5A). Manually curated immune associated signatures representing diverse immune cell types and molecular functions were analyzed by ssGSEA algorithm. LOXL1 high expression group was characterized by escalated infiltration of immunosuppressive components, while LOXL1 low expression group was linked to a richer infiltration of anti-tumor immune cell signatures, including CD8 + T cells and interferon-γ signatures (Fig. 5B). Furthermore, LOXL1 expression was found to be inversely correlated with CD8 + T cells infiltration in several independent cohorts (Fig. 5C and D).
Fig. 5
The distinct landscape of tumor immune microenvironment between patients with high or low LOXL1 expression. (A) Boxplot of 22 immune cell abundance based on deconvolution by CIBERSORT between high and low LOXL1 expression groups in the GSE17536 dataset. Samples were categorized according to the median expression level of LOXL1 gene. The upper and lower ends of the boxes are the 25th and 75th percentiles (interquartile range) respectively. The lines within the boxes represents the median value, and the scattered dots represent outliers. The significance of differences was determined by the Wilcoxon test (ns: not significant, *P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001). (B) The heatmap showed the enrichment level of 25 immune-related gene sets based on single sample gene set enrichment analysis (ssGSEA) analysis in the high and low LOXL1 expression groups. The Student’s t test was used for statistical analysis. (C-D) LOXL1 expression was negatively correlated with the infiltration level of CD8 + T cells in TCGA-COAD cohort (Cor = -0.337, P = 9.45e-09) (C), GSE17536 cohort (Cor = -0.34, P = 3.5e-06), and GSE161158 cohort (Cor = -0.3, P = 1.1e-06) (D). (E) IHC staining of LOXL1 and CD8 in TMA comprising 208 pairs of CRC tissues. Representative IHC staining images from two cases were displayed. (F) Correlation analysis of protein expression between LOXL1 and CD8 in CRC patients using Pearson correlation analysis. H-score represents the value of IOD sum or IOD/Area determined by image pro plus software. (G-I) The heatmap revealed the expression correlation between LOXL1 and multiple chemokines in TCGA-COAD cohort (G), GSE161158 cohort (H), and GSE39582 cohort (I). The Pearson’s correlation coefficient was calculated and demonstrated in each cell, and cells with P value ≥ 0.05 were marked with cross
The protein expression level of LOXL1 and CD8 + T cell infiltration were also investigated by IHC staining in tissue microarray comprising 208 pairs of CRC tissues. Consistently, we found that there existed a negative correlation between LOXL1 expression and CD8 + T cell infiltration (Fig. 5E and F). Since chemokines might facilitate the recruitment of diverse immune cells into tumors (Turner et al. 2014), we sought to determine whether there exists a correlation between LOXL1 expression and chemokines expression. The results revealed that CXCL2, CXCL3, CCL20, CCL25 and CCL28, which played key role in recruiting T lymphocytes to tumor microenvironment (Tosti et al. 2020; Chen, et al. 2020; Gong et al. 2019), were negatively correlated with LOXL1 expression in in dependent cohorts (Fig. 5G-I). Taken together, LOXL1 might be involved in mediating the immuno-suppressive microenvironment leading to poor prognosis of CRC patients.
High expression of LOXL1 predicts poor clinical outcomes of ICBConsidering the immunosuppressive role of LOXL1 in CRC, we wondered whether LOXL1 could predict the clinical efficacy of immunotherapy. The association between LOXL1 levels and previously published gene signatures associated with ICB response (Auslander et al. 2018) were examined in GSE161158 and GSE17536 cohorts. The result revealed a negative correlation between LOXL1 expression and favorable gene signatures associated with ICB response, such as tumor-infiltrating lymphocytes (TILs). Conversely, a positive correlation with the presence of immune-suppressive myeloid-derived suppressor cells which might suppress immune response was observed (Fig. 6A and B). The IPS have been reported to potently predict patients’ response to ICB based on immunogenicity (Charoentong et al. 2017). We then explore the correlation between LOXL1 expression and IPS in CRC patients. We found that as LOXL1 expression elevated, EC (effector cells) score, CP (immune checkpoint) score, and IPS (immunophenoscore) declined, while the SC (suppressor cells) score increased in two independent cohorts (Fig. 6C and D). The TIDE approach was also utilized and revealed a significant positive correlation between the levels of LOXL1 expression and the TIDE scores (Fig. 6E). In addition, LOXL1 expression was found to be substantially elevated in the group of patients who did not respond to treatment compared to those who did, as indicated by the TIDE analysis (Wilcoxon test, P < 0.001) (Fig. 6F), suggesting the potential of LOXL1 as a predictive biomarker for the effectiveness of ICB therapy.
Fig. 6
The expression of LOXL1 could predict patients’ responsiveness to immune checkpoint blockade (ICB) treatment and prognosis. (A) Pearson correlation analysis was used to determine the correlation between LOXL1 expression and key immune modulators in GSE161158 cohort. The square color indicates correlation coefficients, and the square size represents the statistical P value, with larger size indicating greater statistical significance. (B) Violin plots comparing the expression levels of key immune modulators between the high and low LOXL1 expression tumors in GSE17536 cohort. The statistical analysis was determined by Wilcoxon test. (C) Chord diagram illustrated the association between LOXL1 expression and MHC molecular (MHC), effector cell (EC), immunosuppressive cell (SC), immunophenoscore (IPS) score in GSE161158 cohort. Colors indicate correlation coefficients. Pearson correlation analysis was used for statistical analysis (ns: not significant, *P < 0.05; **P < 0.01; ***P < 0.001, **** P < 0.0001). (D) The correlation between LOXL1 expression and MHC, EC, SC, IPS score in GSE17536 cohort. (E) LOXL1 expression was positively associated with tumor immune dysfunction and exclusion (TIDE) scores representing ICB responsiveness in GSE17536 cohort and GSE29621 cohort. (F) Differences in LOXL1 expression between putative immunotherapeutic responders and non-responders from TIDE in GSE17536 cohort and GSE29621 cohort. (G) Comparison of LOXL1 expression between patients responding or not responding to anti-PD-L1 blockade immunotherapy in IMvigor210 cohort. Wilcoxon test was used for statistical analysis. (H) Kaplan–Meier curve showed that high LOXL1 expression predicted worse survival outcome in IMvigor210 cohort (log-rank test, P = 0.04). (I) Stacked bar graph indicated that LOXL1 expression was significantly associated with poor treatment response to anti-PD-L1 immunotherapy in IMvigor210 cohort. (chi-square test, P-value = 1.13E-3). CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease
To verify whether LOXL1 could be used as a biomarker, an anti-PD-L1 immunotherapy cohort (IMvigor210) with detailed clinical data was adopted. Interestingly, a lower LOXL1 expression level was associated with enhanced responses to anti-PD-L1 therapy, in contrast to those with higher LOXL1 levels who exhibited negligible response (Wilcoxon test, P = 7.23e-04) (Fig. 6G). A Kaplan–Meier survival analysis further established a significant inverse relationship between LOXL1 expression and survival outcomes following anti-PD-L1 treatment (Log- rank test, P < 0.05) (Fig. 6H). Moreover, our analysis revealed a markedly higher occurrence of complete or partial response (CR/PR) in patients with lower LOXL1 expression (35%) compared to those with higher expression (14%), as confirmed by a Chi-square test (P = 1.01e-03) (Fig. 6I). Collectively, these results suggested the predictive role for LOXL1 in clinical benefit of ICB.
Overview of LOXL1 in pan-cancerTo examine the prognostic value and immune regulatory influence of LOXL1 across pan-cancer, we analyzed its expression in 24 solid tumor types from TCGA database. The results, which is sourced from UALCAN, revealed that LOXL1 was dysregulated in 13 types of cancers, with 12 of them showing significant upregulation (Fig. 7A). Analysis of data from multiple databases, including UALCAN, TIMER 2.0, and TNMplot, consistently indicates an upregulation of LOXL1 expression in colorectal cancer compared to normal tissue (Supplementary Fig. 4A-D). Metastatic tumors displayed elevated expression of LOXL1 compared with primary tumors and normal tissues across several cancers from TNM plotter database (Supplementary Fig. 5A). Additionally, LOXL1 expression was found increased with tumor progression from early stage to advanced stage (Supplementary Fig. 5B). Kaplan–Meier analysis was then performed and the results demonstrated that high LOXL1 expression predicted shorter OS and DFS in a variety of cancer types (Fig. 7B, Supplementary Fig. 5C). The genetic alterations of LOXL1 in pan-cancer were also assessed using cBioPortal database. The most common alteration type of LOXL1 was “amplification” in the majority of tumors, and multiple types of LOXL1 mutations were observed (Supplementary Fig. 5D and E).
Fig. 7
Role of LOXL1 in pan-cancer. (A) The expression levels of LOXL1 in tumor tissues and corresponding normal tissues from UALCAN database. Student’s t test was used for statistical analysis (ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001). (B) The upper panel showed the survival map using the online tool of Gene Expression Profiling Interactive Analysis (GEPIA2). The Kaplan–Meier survival plots in the lower panel indicated that high LOXL1 expression correlated with poor survival outcome in different kinds of cancer (COAD, GBM, KIRC, LGG, LUAD and SARC). (C) Correlation of LOXL1 expression with the infiltration level of immune cells in COAD, BRCA and HNSC. (D) Pearson correlation was analyzed between LOXL1 expression and the infiltration of cancer-associated fibroblast (CAF) (left panel) and endothelial cell (EC) (right panel) based on the EPIC, MCPCOUNTEER, XCELL and TIDE algorithms. (E) Comparison of LOXL1 expression among different immune infiltration subtypes in multiple cancers from the Tumor–Immune System Interactions and Drug Bank (TISIDB) database. (C1, wound healing; C2, IFN-gamma dominant; C3, inflammatory; C4, lymphocyte depleted; C5, immunologically quiet; and C6, TGF-b dominant)
Additionally, LOXL1 expression and immune cell infiltration were correlated using TIMER2.0 database. Negative correlation was found between LOXL1 expression and the infiltration abundance of anti-tumor immune cells such as CD8 + T cell, CD4 + T cell, B cell, and dendritic cell in COAD, BRCA, and HNSC. Conversely, LOXL1 expression positively correlated with a series of immunosuppressive cells including Tregs, and macrophage (Fig. 7C). Remarkably, cancer-associated fibroblast and endothelial cells, which have been reported to promote tumor progression (Chen and Song 2019; Yang et al. 2021), exhibited strong positive correlations with LOXL1 expression in the majority of cancers (Fig. 7D), indicating the immune-regulatory role of LOXL1 in cancers. Additionally, using the TISIDB database, we explored LOXL1 expression across different immune subtypes in various cancers. Tumors were categorized into six immune subtypes: C1 (wound healing), C2 (IFN-gamma dominant), C3 (inflammatory), C4 (lymphocyte depleted), C5 (immunologically quiet), and C6 (TGF-β dominant) (Thorsson et al. 2018). Our findings revealed a marked enrichment of LOXL1 in wound healing and TGF-β dominant subtypes and a downregulated in IFN-γ dominant subtypes in cancers including colorectal (Fig. 7E), suggesting LOXL1’s immunosuppressive role in pan-cancer.
留言 (0)