Bioinformatics analysis
In this study, the BIOGRID database (https://thebiogrid.org/) was used to screen the proteins that interacted with NEDD4L (Oughtred et al. 2021). The Ubibrower database (http://ubibrowser.bio-it.cn/ubibrowser/) was used to screen potential ubiquitinated E3 ligases of TFRC (Li et al. 2017). In the m6A analysis, RM2Target database (http://rm2target.canceromics.org/#/citation) had been employed for analyze the RNA methylation correlation analysis of potential binding of NEDD4L (Bao et al. 2023), and the SRAMP database (http://www.cuilab.cn/sramp) had been employed to predict the RNA binding targets of NEDD4L and draw the secondary structure (Zhou et al. 2016).
Animals and middle cerebral artery occlusion (MCAO) model
All C57BL/6 mice, aged 8–10 weeks, were purchased from Beijing Charles River Laboratory Animal Company and housed in a pathogen-free facility under a 12-h light/dark cycle with ad libitum access to food and water. All animal studies were performed in accordance with the Ethical Guidelines for the Use and Care of Laboratory Animals and were approved by the Animal Ethics Committee of Sichuan Provincial People’s Hospital.
All mice were fasted and water deprived for 12 h. MCAO model was established by internal carotid artery occlusion method. The mice were intraperitoneally injected with pentobarbital at a dosage of 60 mg/kg to induce anesthesia and subsequently positioned in the prone stance for stabilization. The neck was disinfected, and a 1.5-cm incision was cut located at the midpoint of the cervical region. The subcutaneous tissue, glands, and muscles were separated, and the right common carotid artery and internal carotid artery were exposed.
A knot was tied at both ends of the external carotid artery, a small incision was cut between the two knots, a thread plug was inserted, and the external carotid artery was pulled, so that the thread plug slowly entered the internal carotid artery about 10 mm or so if slight resistance was encountered, the entry was stopped. The thread plug was securely fastened, followed by the suturing of the incision in a layered manner. Furthermore, appropriate disinfection measures were implemented on the thread plug to effectively mitigate any potential risk of infection. Mice underwent reperfusion after 90 min of occlusion. In the sham operation group, the operation was the same as tMCAO, but the suture plug only penetrated about 5 mm, without causing cerebral ischemia.
Construction of NEDD4L knockout mice
NEDD4L conditional knockout mice were purchased from Guangzhou Cyagen Biosciences. To generate NEDD4L knockout mice, we designed a guide RNA targeting exon 7 of NEDD4L and cloned it into the PX459 vector. Mouse embryonic stem cells were transfected with this construct, and single cell clones were isolated. These clones were screened by PCR amplification and sequencing of the target region to identify mutant alleles. Southern blot analysis further validated NEDD4L disruption in positive clones. The appropriately directed embryonic stem cells were subsequently introduced into mouse blastocysts and implanted into surrogate mothers to generate chimeric progeny. Chimeric mice were bred with wildtype mice to achieve germline transmission of the NEDD4L mutation. Intercrosses between NEDD4L heterozygotes resulted in homozygous knockout mice. Genotyping confirmed the absence of wildtype NEDD4L alleles, and western blotting verified deletion of NEDD4L protein in the homozygous mice (Gao et al. 2021).
In vitro cell culture, oxygen–glucose deprivation/reperfusion (OGD/R)
The OGD/R assay was utilized for the in vitro experiments. In brief, mouse hippocampal (HT-22) cells (obtained from Procell Co, LTD) was exposed to 1% oxygen in glucose-free DMEM at 37 ℃ for 4 h for OGD. The O2 level in the medium was guaranteed to equilibrate to 1% hypoxia within 2 h under hypoxic conditions. To ensure the effect of hypoxia, an adequate amount of sugar-free DMEM was added to the dishes of the hypoxia workstation for 4 h before OGD. In addition, oxygen–glucose deprivation manipulations were performed in a hypoxic workstation in this study to eliminate oxygen exposure. Throughout this time period, HT-22 cells were cultured in a complete medium and exposed to a humid CO2 and 95% air for 8 h prior to collection same washing and medium modifications but were consistently maintained at a temperature of 37 ℃ in a complete medium with 5% CO2 and 95% air. The melatonin was initially dissolved in pure ethanol and subsequently diluted to achieve the desired concentrations in the basal medium. In this study, 20 μM and 40 μM of Mel were added 30 min before OGD/R treatment and then treated.
2,3,5-triphenyl tetrazolium chloride (TTC) assay
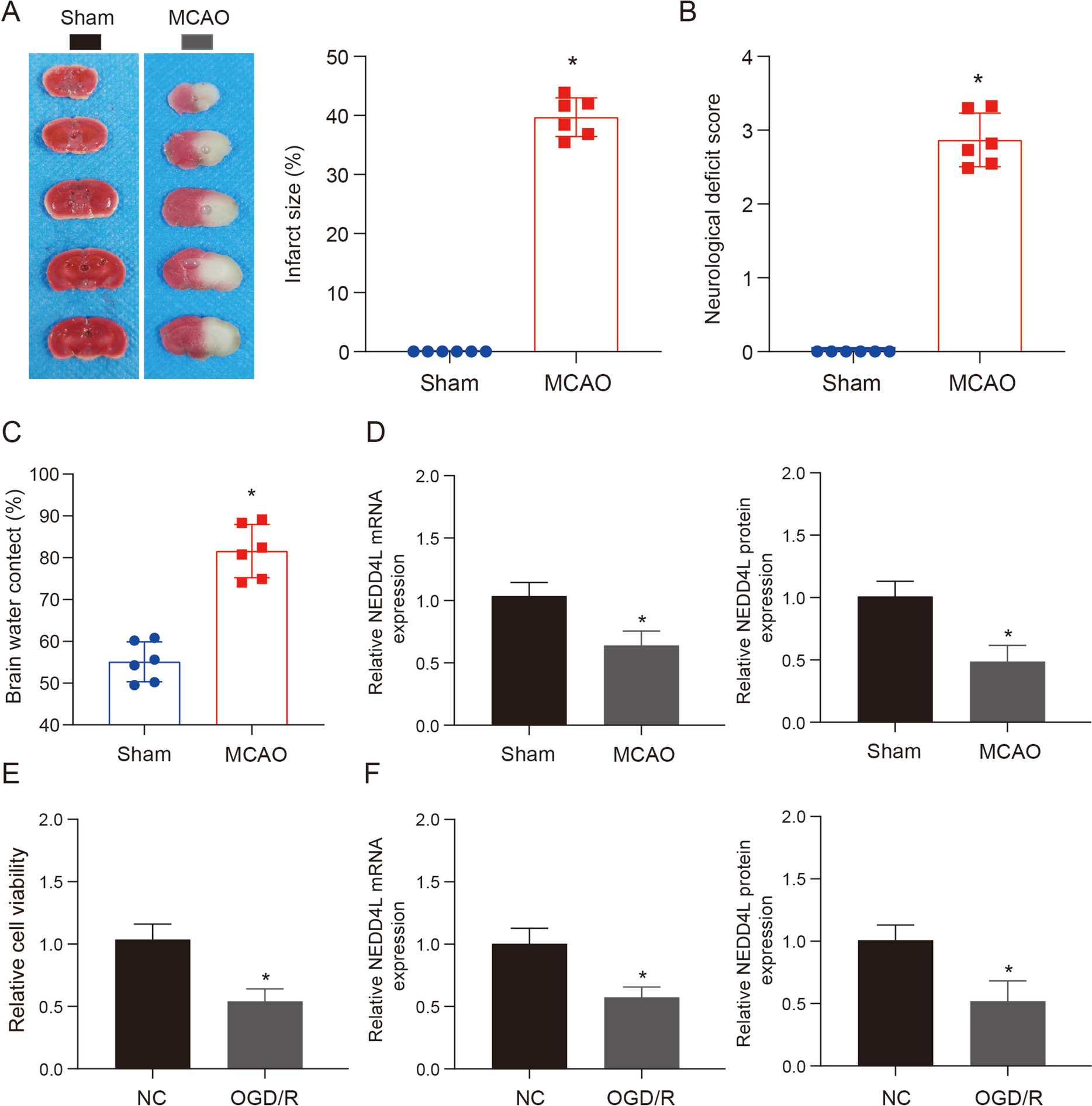
After MCAO operation for 48 h, slowly separate the skull base tissue to obtain complete brain tissue, put it on the mold, and cut it into four coronal slices, each about 2 mm thick. Immerse the brain slices in TTC solution, incubate them at 37 ℃ for 30 min, take out the brain slices, and fix them with paraformaldehyde for 24 h. Scan and image them with a scanner. Normal brain tissues are red areas, ischemic brain tissues are white areas, and the volume of cerebral infarction is measured with Image J software.
Assessment of stroke injury
The evaluation of each mouse was conducted by three examiners who were blinded to the treatment regimen. Scores range from 0 (no dyskinesia) to 4 (severe). After evaluating neurological deficits, the infarct area was photographed and the wet weight of the brain slices was quantified. The slices were then dried at 105 ℃ for 48 hours to determine their water content. Brain water content was calculated using the formula (wet weight - dry weight) / wet weight × 100%.
Cell viability assay
The viability of cells in each treatment group was evaluated using the Cell Counting Kit-8 (CCK-8) assay in this study The supplier’s instructions were meticulously adhered to in order to precisely execute the designated procedures, and subsequent measurement of absorbance at 450 nm was performed for each treatment group.
The proliferation of HT-22 cells was assessed using 5-ethynyl-2′-deoxyuridine (EdU) staining. Various treatments were administered to HT-22 cells, followed by a 2-h incubation with EdU (20 mmol). Subsequently, the cells were treated with 4% paraformaldehyde and left to fix at ambient temperature for a duration of 20 min. The presence of EDU-positive cells in groups was analyzed utilizing Image J.
Cell transfection
SiRNA (si-NEDD4L, si-METTL3) and overexpressed vectors (NEDD4L, TFRC) were synthesized from Genepharma company (Shanghai, China). The HT-22 cells were planed 24 h before transfection, and 20 ~ 100 nmol siRNA was transfected according to cell tolerance and transfection efficiency. The reaction system was 4 mL serum-free medium, cultured for 8 h, then replaced with fresh medium, and continued culture for 48 h.
Real-time fluorescence quantitative PCR (qRT-PCR)
Total RNA of samples was extracted by TRIzol method, and cDNA reverse transcription was performed according to Transcriptor First Strand cDNA Synthesis Kit. β-Actin was used as internal reference. Fast Start Universal SYBR Green Master (Rox) was used for quantitative PCR amplification. PCR reaction system was 20 μL, and the reaction conditions were as follows: an initial denaturation at 95 ℃ for a duration of 2 min, followed by subsequent denaturation at 95 ℃ for a period of 15 s. The annealing process was conducted at a temperature of 60 ℃ for a duration of 30 s. This cycle was repeated for a total number of 40 times. ABI 7500 SDS V1.4 software was used to analyze the data, and 2−ΔΔCT was used for relative quantification. All primer sequences were shown in Supplementary Table 1.
Western blotting (WB)
The BCA method was employed to quantify the protein concentration extracted from RIPA lysate. Polyacrylamide gel was used for electrophoresis. After electrophoresis, the membrane was wet transferred to 0.45 μm PVDF membrane, reconstituted with 5% low-fat milk and incubated for 2 h, followed by overnight incubation at a temperature of 4 ℃ in the presence of primary antibody, washed with TBST (3 times, 10 min/time), selected HRP labeled IgG from the same source as the primary antibody (anti-TFRC, TFRC, 1:1000; anti-NEDD4L, NEDD4L, 1:1000; anti-ACSL4, ACSL4, 1:2000; anti-GPX4, GPX4, 1:2000; anti-FSP1, ab197896, 1:1000; anti-α-tubulin, ab7291, 1:8000) and incubated at room temperature for 1.5 h, TBST washed the membrane (3 times, 10 min/ time), film exposure, development, and fixing.
Hematoxylin and eosin (H&E) and Nissl staining
Brains were fixed overnight in 4% paraformaldehyde, embedded in paraffin, and serially sectioned, deparaffinized, and hydrated. The tissue samples underwent routine histological examination using a light microscope following staining with either H&E or 0.1% cresyl violet.
RNA degradation assay
The cells were initially placed in 6 wells of cell plates with a density of 4 × 105 cells per well. The si-NC group and si-METTL3 were created using the same transfection methods mentioned earlier. After incubating for 48 h, actinomycin was added to inhibit RNA synthesis, following established procedures. Additionally, TRIzol was used to extract total RNA at three time points: immediately and after 6 or 12-h period. Subsequently, cDNA was synthesized through reverse transcription using cDNA synthesis kit. Finally, qRT-PCR was used to assess the expression of NEDD4L mRNA.
TFRC ubiquitination assay
To identify whether TFRC is the substrate of NEDD4L in cells, cell lines were transiently transfected with pcDNA-Flag-NEDD4L plasmid and pcDNA-His-Ub plasmid, respectively, and the cells were lysed 48 h after transfection. The cells were pre-treated with 10 μM MG132 for 4 h prior to sample collection; 1 mg of total protein was used for co-immunoprecipitation, Flag-TFRC was pulled down using anti-Flag affinity gel, and TFRC ubiquitination was detected using anti-Ub antibody in western blot.
Measurement of lipid ROS
The HT-22 cells were cultured in 96-well plates with glass bottoms at a seeding density of 3000 cells per well prior to treatment. Subsequently, the cells underwent treatment and were stained simultaneously using BODIPY-C11 dye and DAPI. Following removal of any excess dye through washing, digital images of the stained cells were captured using an Olympus laser scanning confocal microscope equipped with a 60 × objective lens.
Glutathione measurements
The HT-22 cells were cultured in 6-well plates at a seeding density of 5 × 104 cells per well. After subjecting the cells to the indicated treatments for 48 hours, total intracellular glutathione (GSH) was collected and processed using an assay kit obtained from Nanjing, China, in accordance with the manufacturer's instructions.
Statistical analysis
The data was analyzed using SPSS 20.0 software, and the experimental results were presented as mean ± standard deviation (mean ± SD). The one-way analysis of variance (ANOVA) was employed to compare three or more groups, followed by the least significant difference (LSD) test for subsequent pairwise comparisons. Independent sample t test was used for comparing two groups. The observed difference exhibited statistical significance at P < 0.05 level.

留言 (0)