
Remember me





Hypertension, which is a serious chronic disease, is a key risk factor for cardiovascular adverse events. Vascular remodeling is the pathological basis of hypertension and includes histological or functional changes, such as thickening of the vessel wall, an increase in the vessel wall thickness/lumen ratio, an increase in the extracellular matrix and the phenotypic transformation of vascular smooth muscle cells (VSMCs) [1]. Many studies have shown that improving vascular remodeling can reduce blood pressure (BP) in animal models of hypertension.
Specialized pro-resolving lipid mediators (SPMs) are a kind of endogenous small molecular substances with anti-inflammatory and pro-inflammatory resolution effects that are produced by the metabolism of Ω-3 and Ω-6 polyunsaturated fatty acids (PUFAs) [2,3]. Some clinical studies have shown that supplementation with PUFAs can reduce BP in hypertensive patients and reduce the risk of hypertension in normal individuals [4–6]. As metabolites of PUFAs, SPMs may have stronger biological effects and antihypertensive effects. Resolvin D1 (RvD1) is an SPM derived from docosahexaenoic acids that has significant anti-inflammatory and pro-inflammatory resolution effects [2,3]. RvD1 can inhibit inflammation, reduce cardiomyocyte apoptosis and improve myocardial injury [7]. It can also resolve inflammation in the spleen and ventricle after myocardial infarction and improve cardiac function [8]. A recent study showed that RvD1 could prevent Ang II-induced hypertension [9]. However, the mechanism by which RvD1 improves hypertension is unclear.
Ras homolog gene family member A (RhoA) is a GTP-binding protein, and its downstream signaling pathways, including the mitogen-activated protein kinase (MAPK)-signaling pathway, can regulate cell proliferation, migration, differentiation and apoptosis [10,11]. During hypertension, the RhoA-related signaling pathway mainly affects the proliferation, migration and differentiation of VSMCs, thus affecting hypertension [12–14]. Interestingly, RvD1 has been shown to inhibit the activation of RhoA [15].
Therefore, we hypothesize that RvD1 plays an important role in vascular remodeling in hypertension by regulating the proliferation, migration and phenotypic transformation of VSMCs by inhibiting the RhoA-related signaling pathway.
MATERIALS AND METHODS ReagentsAng II was purchased from Enzo Life Sciences (ALX-151–039-M025, Farmingdale, New York, USA). RvD1 was purchased from Cayman Chemical (10012554, Ann Arbor, Michigan, USA). The RhoA agonist U46619 [16] was purchased from ApexBio Technology (B6890, Houston, Texas, USA).
AnimalsAll animal care and experimental procedures were in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication, revised 2011) and were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University (Wuhan, China).
Male C57BL/6J mice between 8 and 9 weeks old and weighing 23–25 g were obtained from GemPharmatech (Nanjing, China). The mice were maintained in a standard laboratory at the Cardiovascular Research Institute of Wuhan University. The animals were kept in cages with light/dark cycles of 12 h and had free access to food and water. After acclimating to the environment for 2 weeks, the animals were used for the experiments.
Ang II-induced hypertension model and treatmentsThe mice were anesthetized by an intraperitoneal injection (i.p.) of 3% amobarbital at a dose of 70 mg/kg. The hair on the left shoulder blade was shaved, a small incision was made in the middle of the shoulder blade, and the skin and fascia were separated by blunt dissection with hemostatic forceps. Then, an osmotic pump (Alzet, model 2004) was placed, and the wound was sutured and disinfected. The mice were allowed to wake up in the incubator before going back to their cages. For control mice, the wound was closed without implanting the pump. The mice were infused with Ang II (750 ng/min/kg) for 28 days by pumps, and saline or RvD1 (2 μg/kg/day, i.p.) was administered daily starting on the day of the surgery and lasting until the animals were euthanized. After 28 days, the mice were euthanized, and aortic tissue was removed for follow-up biological analysis.
Blood pressure measurementA carotid catheter-calibrated tail-cuff system (CODA, Kent Scientific, Torrington, Connecticut, USA) was used. Every other day for 1 week prior to pump implantation, the mice were placed on the scaffold, the BP meter was activated for 10 min to acclimate, and the animals were then allowed to return to the cage. The day before surgery, the mice were tethered to the scaffold, and their tails were heated on a heating pad for 20 min before 15 cycles basal SBP, DBP, mean BP (MBP) and heart rate (HR) measurements was performed. This procedure was repeated at 7, 14 and 28 days after Ang II infusion.
Histological analysisAortic tissue was separated. After being fixed with 4% paraformaldehyde for 3 days, the tissue was embedded in paraffin and sliced into approximately 5-μm-thick sections. Aortic media thickness was analyzed by hematoxylin–eosin staining, and Masson trichrome staining was used to examine collagen deposition. The sections were then visualized, and images were captured by light microscopy (Olympus, Japan). Media thickness and the area of collagen deposition were measured using a quantitative digital image analysis system (ImagePro Plus, version 6.0).
Immunofluorescence analysisThe aortic tissue was fixed with 4% paraformaldehyde for 3 days and embedded in paraffin wax. The samples were sliced into approximately 5-μm-thick sections. After 5 min of high-pressure antigen repair (sodium citrate buffer, 100×, pH 6.0), the sections were incubated in PBS containing 10% fetal bovine serum (FBS) for 60 min. Then, the aortic sections were incubated overnight at 4 °C with the following primary antibodies: antialpha smooth muscle actin (α-SMA) (ab5694, Abcam, Cambridge, UK), antismooth muscle protein 22-α (SM22α) (GB11366, Servicebio, Wuhan, China), and antiosteopontin (OPN) (22952-1-AP, Proteintech, Wuhan, China). The sections were washed with PBS and incubated with appropriate secondary antibodies at 37 °C for 1 h. The nuclei were stained with 4,6 diamino-2-phenylindole (DAPI). Images were collected by a fluorescence microscope (Olympus, Japan) and DP2-BSW software (Version 2.2) and analyzed in a blinded manner by Image-Pro Plus (Version 6.0) software.
Cell culture and treatmentsVSMCs were extracted from the aortic tissue of 4-week-old rats. The cells were cultured in Dulbecco's modified eagle medium/nutrient mixture F-12 (DMEM/F12) culture medium (SH30023.01, Cytiva) supplemented with 10% FBS (10099-141, Gibco, Grand Island, New York, USA) at 37 °C in a humidified 5% CO2 incubator. The culture medium was refreshed every 3 days. VSMCs were used at passages 3–7. The cells were exposed to Ang II (1000 nmol/l) or U46619 (100 nmol/l) for 24 h. Prior to Ang II treatment, VSMCs were pretreated with RvD1 (1, 10, and 100 nmol/l) for 30 min.
Cell proliferation assessmentCell proliferation was measured using Cell Counting Kit-8 (ANT181, AntGene, Wuhan, China) and EdU imaging kits (K1075, ApexBio Technology) according to the manufacturer's instructions.
Wound healing assayVSMCs were seeded into 6-well plates, grown to fill with wells, and incubated in FBS-free medium for 24 h before a sterile pipette tip was used to wound the cells. After being rinsed with PBS, the cells were treated with Ang II, U46619 and RvD1. The cells were observed under a light microscope (Olympus, Japan) at 0 and 24 h. Different images were randomly collected, the area of cell migration was determined, and the rate of cell migration was calculated by Image-Pro Plus (Version 6.0) software.
Western blottingAortic tissue and VSMCs were lysed in RIPA lysis buffer containing protease and phosphatase inhibitors. The samples were further homogenized by ultrasound and then centrifuged at 12 000g for 30 min. Total protein was collected, and the concentration was determined with a BCA protein assay kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The protein concentration was analyzed after normalization. The proteins (50 μg) were separated by 10% SDS–PAGE and transferred to Immobilon-FL polyvinylidene fluoride (PVDF) membranes (IPFL00010, Merck Millipore Ltd, Darmstadt, Germany). The membranes were blocked with 5% nonfat milk at room temperature for 1 h and incubated overnight at 4 °C on a shaker with the following primary antibodies: antiα-SMA (ab5694, Abcam), anti-SM22α (GB11366, Servicebio), anti-OPN (22952-1-AP, Proteintech), antiproliferating cell nuclear antigen (PCNA) (GTX100539, GeneTex, Irvine, California, USA), anti-RhoA (10749-1-AP, Proteintech), antiphospho-p44/42 MAPK (P-ERK1/2) (4370, Cell Signaling Technology, Danvers, Massachusetts, USA), antip44/42 MAPK (T-ERK1/2) (4695, Cell Signaling Technology), antiphospho-SAPK/JNK (P-JNK1/2) (4668, Cell Signaling Technology), anti-SAPK/JNK (T-JNK1/2) (9258, Cell Signaling Technology), antiphospho-p38 MAPK (P-P38) (4511, Cell Signaling Technology), antip38 MAPK (T-P38) (8690, Cell Signaling Technology), and antiglyceraldehyde-3-phosphate dehydrogenase (GAPDH) (AC027, Abclonal, Wuhan, China). In next day, the membranes were treated with goat antirabbit IgG (#7074, Cell Signaling Technology) or goat antimouse IgG (#7076, Cell Signaling Technology) secondary antibodies at room temperature for 1 h in the dark. After being washed three times in the dark, the membranes were scanned to measure the band intensities by an Odyssey Imaging System (LI-COR Biosciences, Lincoln, USA). The expression levels of the target proteins were normalized to GAPDH.
Statistical analysisAll data are expressed as the mean ± standard deviation (SD) and were analyzed by GraphPad Prism 7 software. Differences among groups were determined by one-way ANOVA or two-way ANOVA followed by Tukey's post hoc tests. P less than 0.05 was considered statistically significant.
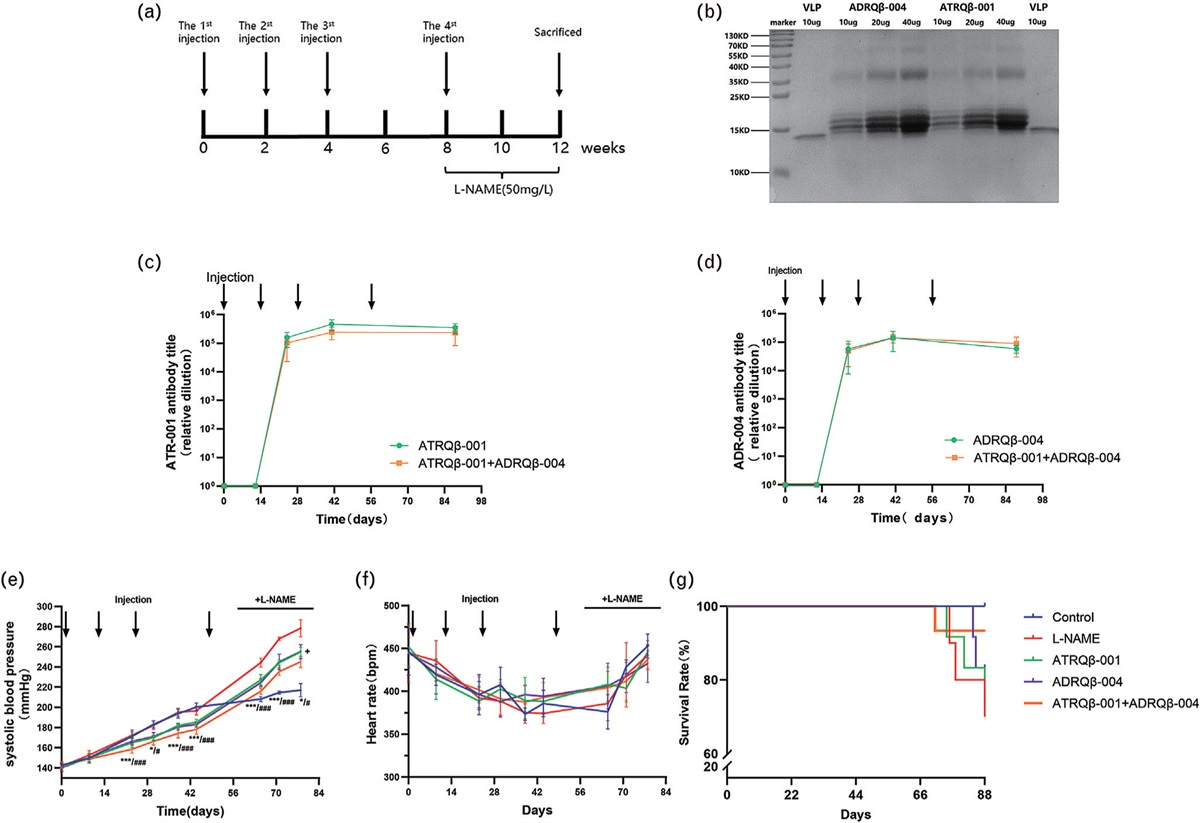
RESULTS Resolvin D1 reduced Ang II-induced hypertension in miceTo clarify the role of RvD1 in hypertension, we established an Ang II-induced hypertension mouse model. The experimental protocol is shown in Fig. 1a. We measured BP once per week for 4 weeks after the mice were implanted with an osmotic pump filled with Ang II. We analyzed BP data measured at the same heart rate and found that the group treated with only Ang II had significantly higher SBP and DBP than the control group, and this increase in BP was suppressed by treatment with RvD1 (Fig. 1b–d). Apparently, RvD1 could reduce Ang II-induced hypertension in mice.
 FIGURE 1:
FIGURE 1: The effects of resolvin D1 in Ang II-induced hypertension model in mice. (a) The experimental protocol. (b–d) SBP and DBP of the mice measured at similar HR once a week for 4 weeks after the mice were placed an osmotic pump prefilled with Ang II (n = 10–12) (SBP, DBP and HR was analyzed by two-way ANOVA followed by Tukey's post hoc tests). Hematoxylin–eosin (e and f) and Masson (g and h) staining were used for displaying the aortic medium thickness and vascular fibrosis, respectively (n = 5) (scale bar, 50 μm). Data are presented as the mean ± SD. ∗P < 0.05 compared with the Vehicle group, #P < 0.05 compared with the Ang II group. HR, heart rate.
Resolvin D1 inhibited Ang II-induced vascular remodeling in miceWe further examined vascular remodeling, which is the pathological basis of hypertension. Aortic tissues were sectioned and stained with hematoxylin–eosin. The results revealed that RvD1 treatment reduced Ang II-induced thickening of the aortic media (Fig. 1e–f). Masson trichrome staining was used to examine collagen deposition. Similarly, after Ang II treatment, fibrosis in the aorta was significantly aggravated, and RvD1 effectively reduced collagen deposition and improved vascular fibrosis (Fig. 1g–h).
In addition to morphological changes, phenotypic transformation of VSMCs was examined. Immunofluorescence was used to visualize changes in aortic tissues. We found that α-SMA and SM22α, which are markers of contractile VSMCs, were significantly downregulated, whereas OPN, which is a marker of synthetic VSMCs, was significantly upregulated, and these changes were reversed by RvD1 (Fig. 2a–d). To further verify this result, we examined the expression of related proteins in aortic tissue by Western blotting. As expected, the results were consistent with the immunofluorescence analysis (Fig. 2e–h).
 FIGURE 2:
FIGURE 2: Resolvin D1 inhibited Ang II-induced phenotypic transformation of vascular smooth muscle cells in aortic. Immunofluorescence showed the expression of α-SMA, SM22α and OPN (a) in the heart and quantitative comparison among different groups (b–d) (n = 5) (scale bar, 50 μm). Protein expression of α-SMA, SM22α and OPN was tested by Western blotting (e). GAPDH was used as internal control and quantification of the α-SMA (f), SM22α (g) and OPN (h) level in the indicated groups (n = 4). Data are presented as the mean ± SD. ∗P < 0.05 compared with the Vehicle group, #P < 0.05 compared with the Ang II group.
Resolvin D1 inhibited the proliferation and migration of vascular smooth muscle cells after Ang II stimulation in vitroTo clarify the mechanism by which RvD1 improves Ang II-induced vascular remodeling, we examined the cellular function of VSMCs. We treated VSMCs with different concentrations of RvD1, and cell proliferation was determined by the Cell Counting Kit-8 assay. The results showed that 100 nmol/l RvD1 inhibited the proliferation of VSMCs after Ang II stimulation (Fig. 3a). Therefore, we used this concentration to stimulate the cells in subsequent experiments. EdU imaging kits showed that more proliferating cells were labeled after Ang II stimulation, and this effect was inhibited by treatment with RvD1 (Fig. 3b and c). In addition to cell proliferation, migration was also evaluated by using a wound healing assay. The ability of cells to migrate was significantly enhanced after stimulation with Ang II, and this effect was decreased by RvD1 treatment (Fig. 3d and e).
 FIGURE 3:
FIGURE 3: Resolvin D1 inhibited the proliferation and migration of VSMCs after Ang II stimulation. (a) Different doses of RvD1 (1 nmol/l, 10 nmol/l, 100 nmol/l) were used to treat VSMCs and determined cell proliferation using the Cell Counting Kit-8 (n = 10). (b and c) EdU Imaging Kits (Cy3) were used to display and quantify proliferating cells for comparison between groups (n = 7–8) (scale bar, 50 μm). (d) Wound healing assay recorded the migration of VSMCs after 24 h of different stimulation (scale bar, 500 μm). (e) The rate of cell migration was calculated and compared (n = 4). Data are presented as the mean ± SD. ∗P < 0.05 compared with the PBS group, #P < 0.05 compared with the Ang II group.
Resolvin D1 inhibited the phenotypic transformation of vascular smooth muscle cells after Ang II stimulation in vitroWe confirmed that RvD1 attenuated Ang II-induced phenotypic transformation in VSMCs in vivo. Relevant parameters were also examined in vitro. After Ang II stimulation, the expression of α-SMA and SM22α was decreased, whereas that of OPN and PCNA was increased, and these effects were reversed by RvD1 (Fig. 4a–e).
 FIGURE 4:
FIGURE 4: Resolvin D1 inhibited the phenotypic transformation of VSMCs after Ang II stimulation. Protein expression of α-SMA, SM22α, OPN and PCNA was tested by Western blotting (a). GAPDH was used as internal control and quantification of the α-SMA (b), SM22α (c), OPN (d) and PCNA (e) level in the indicated groups (n = 4). Data are presented as the mean ± SD. ∗P < 0.05 compared with the PBS group, #P < 0.05 compared with the Ang II group.
Resolvin D1 attenuated the proliferation, migration and phenotypic transformation of VSMCs after Ang II stimulation by inhibiting the RhoA/MAPK pathwayStudies have shown that RhoA/MAPK can promote the proliferation, migration and differentiation of VSMCs during hypertension [11,17,18]. Therefore, we examined the changes in the expression of this pathway in vitro and in vivo. The in-vivo results showed that the expression of RhoA was significantly increased in the Ang II group, and this effect was inhibited by treatment with RvD1 (Fig. 5a–b). The downstream MAPK signaling pathway also showed the same trend (Fig. 5a,c–e). In vitro, we obtained similar results (Fig. 5f–j).
 FIGURE 5:
FIGURE 5: Resolvin D1 inhibited the expression of RhoA/MAPK pathway after Ang II treatment in vivo/in vitro. (a and f) Protein expression of RhoA/MAPK signaling pathway in vivo/in vitro was tested by Western blotting. GAPDH was used as internal control and quantification of RhoA (b and g). T-ERK, T-JNK and T-P38 were used as internal control and quantification of the P-ERK (c and h), P-JNK (d and i) and P-P38 (e and j) level in the indicated groups, respectively (n = 4). Data are presented as the mean ± SD. ∗P < 0.05 compared with the Vehicle/PBS group, #P < 0.05 compared with the Ang II group.
To further verify whether RvD1 improves vascular remodeling by inhibiting the RhoA/MAPK pathway, we used the RhoA agonist U46619 in vitro. RvD1 reduced Ang II-induced proliferation of VSMCs, the ameliorative effect was abrogated by U46619 (Fig. 6a–c). U46619 also counteracted RvD1-mediated inhibition of the migration of VSMCs (Fig. 6d–e). Similarly, RvD1 upregulated the markers of contractile VSMCs (α-SMA and SM22α) and downregulated the markers of synthetic VSMCs (OPN and PCNA), and these effects could be reversed by U46619 (Fig. 7a–e). Then, we examined the activation of the RhoA/MAPK pathway, and U46619 abolished the inhibitory effect of RvD1 on the activation of the RhoA/MAPK pathway (Fig. 7a,f–i). These results indicated that the RhoA/MAPK pathway could mediate the protective effects of RvD1 against the proliferation, migration and phenotypic transformation of VSMCs.
 FIGURE 6:
FIGURE 6: U46619 reversed the inhibitory effect of resolvin D1 on the proliferation and migration of VSMCs after Ang II stimulation. (a) Cell Counting Kit-8 was used to determine cell proliferation (n = 15). (b and c) EdU Imaging Kits (Cy3) was used to examine the proportion of proliferating cells (n = 6) (scale bar, 50 μm). (d and e) Wound healing assay recorded the migration of VSMCs after 24 h of different stimulation (scale bar, 500 μm), the rate of cell migration was calculated and compared (n = 6). Data are presented as the mean ± SD. ∗P < 0.05 compared with the Ang II group, #P < 0.05 compared with the Ang II+RvD1 group.
 FIGURE 7:
FIGURE 7: U46619 reversed the inhibitory effect of resolvin D1 on the phenotypic transformation and the expression of RhoA/MAPK pathway in VSMCs after Ang II stimulation. (a) Protein expression of α-SMA, SM22α, OPN, PCNA and RhoA/MAPK signaling pathway in vitro was tested by Western blotting. GAPDH was used as internal control and quantification of the α-SMA (b), SM22α (c), OPN (d), PCNA (e), RhoA (f). T-ERK, T-JNK and T-P38 were used as internal control and quantification of the P-ERK (g), P-JNK (h) and P-P38 (i) level in the indicated groups respectively (n = 4). Data are presented as the mean ± SD. ∗P < 0.05 compared with the Ang II group, #P < 0.05 compared with the Ang II+RvD1 group.
DISCUSSIONIn this study, we explored the role of RvD1, an internal metabolite of PUFAs, in hypertension. This study showed that RvD1 treatment could inhibit Ang II-induced hypertension and vascular remodeling. Mechanistically, RvD1 was considered a novel inhibitor of RhoA, and it could inhibit RhoA/MAPK pathway activation to inhibit Ang II-induced proliferation, migration and phenotypic transformation of VSMCs, thereby reducing BP, thickening of the aortic media and vascular fibrosis (Fig. 8). Thus, this study provides compelling evidence supporting a protective effect of RvD1 against hypertension and vascular remodeling. RvD1 may be a new drug for treating hypertension or other vascular remodeling-related diseases.
 FIGURE 8:
FIGURE 8: Resolvin D1 attenuates Ang II-induced hypertension through inhibiting the proliferation, migration and phenotypic transformation of vascular smooth muscle cells via blocking the RhoA/MAPK pathway.
Vascular remodeling is a typical pathological feature of many cardiovascular diseases, such as atherosclerosis, aortic dissection, arterial aneurysm and hypertension. SPMs are endogenous small molecules with anti-inflammatory and pro-inflammatory resolution effects, and an increasing number of SPM-related genetic models and pharmacological experiments have shown that SPMs protect against the occurrence and development of vascular remodeling [19]. The levels of SPMs, including RvD1, resolvin D2, and resolvin E1, are significantly reduced in atherosclerosis [20,21]. Numerous studies have shown that different SPMs, when combined with corresponding receptors, could improve vascular remodeling in atherosclerosis by inhibiting inflammation and oxidative stress, thereby reducing the activation of VSMCs or promoting the transformation of macrophages to repair phenotypes [21–25]. Moreover, SPMs can modulate the phenotype of VSMCs and are correlated with peripheral atherosclerosis [26]. Similarly, in aortic dissection and arterial aneurysm, SPMs alleviated vascular remodeling mainly through anti-inflammatory and pro-inflammatory resolution effects [27–29]. In addition, SPMs could inhibit the proliferation, migration and phenotypic transformation of VSMCs, thus reducing vascular remodeling after arterial injury [30,31].
Previous studies have shown that there is an imbalance between proinflammatory and resolution mediators in hypertension, and low SPM levels may be an indicator of the development of hypertension [32,33]. Resolvin D2 has been proven to improve Ang II-induced hypertension by modulating vascular factors, fibrosis and inflammation to protect cardiovascular structure and function [33]. RvD1 also plays a cardioprotective role in angiotensin II-induced hypertension and cardiac remodeling, but the mechanism by which RvD1 improves hypertension is unclear [9]. In this study, we used RvD1 prophylactically to examine its role in hypertension. This study further confirmed that RvD1 could attenuate Ang II-induced hypertension by inhibiting the proliferation, migration and phenotypic transformation of VSMCs.
The biological activities of SPMs are mediated by their cognate G protein-coupled transmembrane superfamily receptors (GPCRs) [34]. Two specific GPCRs for RvD1 have been identified: formyl peptide receptor 2 (FPR2) and G protein-coupled receptor 32 (GPR32) [34,35]. FPR2 and GPR32 are mainly expressed in immune cells, including polymorphonuclear cells, macrophages and T lymphocytes, and they are also expressed in endothelial cells and VSMCs [35]. As GPR32 has only been shown to be expressed in human cells and its homology has not been determined in mice, the effect of RvD1 on a mouse model could be mediated by FPR2 [35,36]. Although the role of RvD1 has been studied more extensively in immune cells than other cell types, recent evidence suggests that VSMCs express RvD1 receptors, and many direct effects of RvD1 have been observed in the vasculature. Multiple in-vivo and in-vitro models have demonstrated that RvD1 treatment can inhibit the proliferation, migration and phenotypic transformation of VSMCs [30,37–39]. These effects of RvD1 could affect the structure and function of vessels. Vascular remodeling is an important pathophysiological basis of hypertension, and VSMCs play an important role. Therefore, we focused on the effect of RvD1 on the cellular function of VSMCs. Notably, RvD1 may affect hypertension through other factors, such as various immune cells or endothelial cells, which needs to be further explored.
RhoA has been widely studied in hypertension. The role of RhoA-related signaling pathways in regulating cell proliferation, migration, differentiation, and apoptosis in hypertension has been examined by many studies [40,41]. As a downstream target of RhoA, the MAPK signaling pathway plays an important role in this process [18]. RvD1 promotes the targeting and clearance of necroptotic cells by inhibiting RhoA activation [15]. The MAPK signaling pathway can also be blocked by treatment with SPMs, including RvD1, in many diseases [7,42,43]. Similarly, we found RhoA/MAPK pathway was inhibited after the treatment with RvD1 in Ang II-induced hypertensive mice and Ang II-stimulated VSMCs. In addition, the contractility of VSMCs is also a key factor in hypertension. Previous studies have shown that SPMs, including RvD1, can inhibit the constriction of the rat thoracic aorta and human pulmonary artery induced by the thromboxane mimetic U46619 [44]. The contractility of VSMCs is related to cytosolic Ca2+ concentrations and the Ca2+ sensitivity of myofilaments, and it has been well documented that the RhoA-related signaling pathway is involved in this process [17,45]. Therefore, RvD1 improves hypertension not only by improving the cellular function of VSMCs but also by correcting the abnormal contractility of VSMCs.
This study used an Ang II-induced hypertension mouse model and Ang II-stimulated rat VSMCs to examine the role of RvD1 in vascular remodeling, which has led to limitations regarding species. Although the species used in vivo and in vitro are different, similar studies have been recognized and widely used in hypertension-related studies [46,47]. Therefore, our study is reliable. In addition, previous studies have shown that the expression levels of SPMs or their precursors are decreased in hypertensive patients or animal models of hypertension [32,48]. This indicates that the results of the present study are credible and provide a new target and important theoretical basis for the treatment of human hypertension. Of course, we will design further studies to verify the effect of RvD1 on humans.
Our study still has other limitations. First, we selected the dose of RvD1 based on previous studies. The dose-dependent effect of RvD1 on improving hypertension needs to be further examined. In this study, we used RvD1 prophylactically and did not examine its therapeutic effects, which may require further experiments. In addition, although we have established that RvD1 can improve hypertension by regulating the cellular function of VSMCs, it may also act on other cells, such as immune cells and endothelial cells, which is worth exploring.
CONCLUSIONThis study provides direct evidence that RvD1 attenuates Ang II-induced hypertension by inhibiting the proliferation, migration and phenotypic transformation of VSMCs by blocking the RhoA/MAPK pathway. This provides new ideas for the diagnosis and treatment of hypertension.
ACKNOWLEDGEMENTSFunding: this work was supported by grants from the National Natural Science Foundation of China (No. 82270454, 82100292 and 82070436) and Excellent Doctoral Program of Zhongnan Hospital of Wuhan University (ZNYB2022001).
Conflicts of interestThere are no conflicts of interest.
REFERENCES 1. Brown IAM, Diederich L, Good ME, DeLalio LJ, Murphy SA, Cortese-Krott MM, et al. Vascular smooth muscle remodeling in conductive and resistance arteries in hypertension. Arterioscler Thromb Vasc Biol 2018; 38:1969–1985. 2. Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol 2008; 153 Suppl 1:S200–S215. 3. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014; 510:92–101. 4. Minihane AM, Armah CK, Miles EA, Madden JM, Clark AB, Caslake MJ, et al. Consumption of fish oil providing amounts of eicosapentaenoic acid and docosahexaenoic acid that can be obtained from the diet reduces blood pressure in adults with systolic hypertension: a retrospective analysis. J Nutr 2016; 146:516–523. 5. Liao J, Xiong Q, Yin Y, Ling Z, Chen S. The effects of fish oil on cardiovascular diseases: systematical evaluation and recent advance. Front Cardiovasc Med 2021; 8:802306. 6. Ni S, Zhong Z, Wei J, Zhou J, Cai L, Yang M, et al. Association between dietary intake of polyunsaturated fatty acid and prevalence of hypertension in U.S. adults: a cross-sectional study using data from NHANES 2009-2016. Hypertens Res 2022; 45:516–526. 7. Wang M, Liu M, Zhang J, Liu J, Ye J, Xu Y, et al. Resolvin D1 protects against sepsis-induced cardiac injury in mice. Biofactors 2020; 46:766–776. 8. Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, et al. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol 2015; 84:24–35. 9. Olivares-Silva F, De Gregorio N, Espitia-Corredor J, Espinoza C, Vivar R, Silva D, et al. Resolvin-D1 attenuation of angiotensin II-induced cardiac inflammation in mice is associated with prevention of cardiac remodeling and hypertension. Biochim Biophys Acta Mol Basis Dis 2021; 1867:166241. 10. Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J 1996; 15:2208–2216. 11. Seccia TM, Rigato M, Ravarotto V, Calo LA. ROCK (RhoA/Rho Kinase) in cardiovascular-renal pathophysiology: a review of new advancements. J Clin Med 2020; 9:1328. 12. Wirth A. Rho kinase and hypertension. Biochim Biophys Acta 2010; 1802:1276–1284. 13. Zou F, Li Y, Zhang S, Zhang J. DP1 (Prostaglandin D(2) Receptor 1) activation protects against vascular remodeling and vascular smooth muscle cell transition to myofibroblasts in angiotensin II-induced hypertension in mice. Hypertension 2022; 79:1203–1215. 14. Arnold C, Demirel E, Feldner A, Genove G, Zhang H, Sticht C, et al. Hypertension-evoked RhoA activity in vascular smooth muscle cells requires RGS5. FASEB J 2018; 32:2021–2035. 15. Gerlach BD, Marinello M, Heinz J, Rymut N, Sansbury BE, Riley CO, et al. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ 2020; 27:525–539. 16. Akbar H, Duan X, Saleem S, Davis AK, Zheng Y. RhoA and Rac1 GTPases differentially regulate agonist-receptor mediated reactive oxygen species generation in platelets. PLoS One 2016; 11:e0163227. 17. Ma J, Li Y, Yang X, Liu K, Zhang X, Zuo X, et al. Signaling pathways in vascular function and hypertension: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther 2023; 8:168. 18. Sawma T, Shaito A, Najm N, Sidani M, Orekhov A, El-Yazbi AF, et al. Role of RhoA and Rho-associated kinase in phenotypic switching of vascular smooth muscle cells: Implications for vascular function. Atherosclerosis 2022; 358:12–28. 19. Diaz Del Campo LS, Rodrigues-Diez R, Salaices M, Briones AM, Garcia-Redondo AB. Specialized pro-resolving lipid mediators: new therapeutic approaches for vascular remodeling. Int J Mol Sci 2022; 23:3592. 20. Elajami TK, Colas RA, Dalli J, Chiang N, Serhan CN, Welty FK. Specialized proresolving lipid mediators in patients with coronary artery disease and their potential for clot remodeling. FASEB J 2016; 30:2792–2801. 21. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun 2016; 7:12859. 22. Petri MH, Laguna-Fernandez A, Gonzalez-Diez M, Paulsson-Berne G, Hansson GK, Back M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc Res 2015; 105:65–74. 23. Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, et al. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res 2016; 119:1030–1038. 24. Back M, Yurdagul A Jr, Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 2019; 16:389–406. 25. Rangarajan S, Orujyan D, Rangchaikul P, Radwan MM. Critical role of inflammation and specialized pro-resolving mediators in the pathogenesis of atherosclerosis. Biomedicines 2022; 10:2829. 26. Ho KJ, Spite M, Owens CD, Lancero H, Kroemer AH, Pande R, et al. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J Pathol 2010; 177:2116–2123. 27. Li B, Yao BC, Guo ZG, Zhang SP, Song YQ, Jiang N, et al. Mechanism of action of resolvin D1 in inhibiting the progression of aortic dissection in mice. Ann Transl Med 2021; 9:1498. 28. Spinosa M, Su G, Salmon MD, Lu G, Cullen JM, Fashandi AZ, et al. Resolvin D1 decreases abdominal aortic aneurysm formation by inhibiting NETosis in a mouse model. J Vasc Surg 2018; 68:93S–103S. 29. Pope NH, Salmon M, Davis JP, Chatterjee A, Su G, Conte MS, et al. D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. FASEB J 2016; 30:4192–4201. 30. Miyahara T, Runge S, Chatterjee A, Chen M, Mottola G, Fitzgerald JM, et al. D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J 2013; 27:2220–2232. 31. Liu G, Gong Y, Zhang R, Piao L, Li X, Liu Q, et al. Resolvin E1 attenuates injury-induced vascular neointimal formation by inhibition of inflammatory responses and vascular smooth muscle cell migration. FASEB J 2018; 32:5413–5425. 32. Yucel H, Ozdemir AT. Low LXA4, RvD1 and RvE1 levels may be an indicator of the development of hypertension. Prostaglandins Leukot Essent Fatty Acids 2021; 174:102365. 33. Diaz Del Campo LS, Garcia-Redondo AB, Rodriguez C, Zaragoza C, Duro-Sanchez S, Palmas F, et al. Resolvin D2 attenuates cardiovascular damage in angiotensin ii-induced hypertension. Hypertension 2023; 80:84–96. 34. Chiang N, Serhan CN. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med 2017; 58:114–129. 35. Pirault J, Back M. Lipoxin and resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Front Pharmacol 2018; 9:1273. 36. Back M, Powell WS, Dahlen SE, Drazen JM, Evans JF, Serhan CN, et al. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br J Pharmacol 2014; 171:3551–3574. 37. Wu B, Mottola G, Chatterjee A, Lance KD, Chen M, Siguenza IO, et al. Perivascular delivery of resolvin D1 inhibits neointimal hyperplasia in a rat model of arterial injury. J Vasc Surg 2017; 65:207.e3–217.e3. 38. Wu B, Werlin EC, Chen M, Mottola G, Chatterjee A, Lance KD, et al. Perivascular delivery of resolvin D1 inhibits neointimal hyperplasia in a rabbit vein graft model. J Vasc Surg 2018; 68: (6 Suppl): 188S.e4–200S.e4. 39. Mottola G, Chatterjee A, Wu B, Chen M, Conte MS. Aspirin-triggered resolvin D1 attenuates PDGF-induced vascular smooth muscle cell migration via the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathway. PLoS One 2017; 12:e0174936. 40. Tang L, Dai F, Liu Y, Yu X, Huang C, Wang Y, et al. RhoA/ROCK signaling regulates smooth muscle phenotypic modulation and vascular remodeling via the JNK pathway and vimentin cytoskeleton. Pharmacol Res 2018; 133:201–212. 41. Qi Y, Liang X, Dai F, Guan H, Sun J, Yao W. RhoA/ROCK pathway activation is regulated by AT1 receptor and participates in smooth muscle migration and dedifferentiation via promoting actin cytoskeleton polymerization. Int J Mol Sci 2020; 21:5398. 42. Zhang J, Wang M, Ye J, Liu J, Xu Y, Wang Z, et al. The anti-inflammatory mediator resolvin E1 protects mice against lipopolysaccharide-induced heart injury. Front Pharmacol 2020; 11:203. 43. Sun S, Wang J, Wang J, Wang F, Yao S, Xia H. Maresin 1 mitigates sepsis-associated acute kidney injury in mice via inhibition of the NF-kappaB/STAT3/MAPK pathways. Front Pharmacol 2019; 10:1323. 44. Jannaway M, Torrens C, Warner JA, Sampson AP. Resolvin E1, resolvin D1 and resolvin D2 inhibit constriction of rat thoracic aorta and human pulmonary artery induced by the thromboxane mimetic U46619. Br J Pharmacol 2018; 175:1100–1108. 45. Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997; 389:990–994. 46. Jiao X, Yu H, Du Z, Li L, Hu C, Du Y, et al. Vascular smooth muscle cells specific deletion of angiopoietin-like protein 8 prevents AngII-promoted hypertension and cardiovascular hypertrophy. Cardiovasc Res 2023; 119:1856–1858. 47. Forrester SJ, Elliott KJ, Kawai T, Obama T, Boyer MJ, Preston KJ, et al. Caveolin-1 deletion prevents hypertensive vascular remodeling induced by angiotensin II. Hypertension 2017; 69:79–86. 48. Edwards-Glenn JM, Fontes MT, Waigi EW, Costa TJ, Maiseyeu A, Webb C, et al. Specialized pro-resolving mediator therapy improves vascular relaxation via formyl peptide-receptor-2. Am J Hypertens 2023.
Comments (0)