Cell lines and growth conditions
We obtained HepG2 and SNU387 HCC cell lines from the American Type Culture Collection (ATCC, Manassas, VA, USA). The Hep3B cell line was obtained from DSZM (Leibniz Institute, Berlin, Germany). These cell lines were cultivated in specific media: Eagle’s Minimum Essential Medium for HepG2 and Hep3B (BE12-6621, Lonza, Basel, Switzerland), and RPMI 1640 Medium for SNU387 (12-167Q, Lonza, Basel, Switzerland). To the media, we added 10% (v/v) fetal calf serum and 1 (w/v) L-glutamine (Lonza). The cells were incubated at 37 °C in an environment with 5% (v/v) CO2 and 100% (v/v) humidity. For the generation and culture of stable transfected cell lines expressing Mimic hsa-miR-423-5p, we employed the same culture media with the addition of 1.5 \(\mu\)g/mL of Puromycin for Hep3B and SNU387, and 2.5 \(\mu\)g/mL of Puromycin for HepG2 to ensure antibiotic selection. As for the HEK293T cell lines, these were acquired from the American Type Culture Collection (Manassas, VA, USA). They were maintained in DMEM media (BE12-604F, Lonza, Basel, Switzerland), supplemented with 10% (v/v) fetal calf serum and 1% (w/v) L-glutamine (Lonza).
Transient transfection of HCC cells with miRNA 423-5p mimic and inhibitor
To clarify the specific role of miRNA 423-5p, HCC cells at 80% of confluence were transfected with miRNA 423-5p mimic and its inhibitor. The day before transfection, cells were trypsinized and seeded in an appropriate medium without antibiotics in 12-well plates. MiRIDIAN miR-423-5p mimic, miRIDIAN miR-423-5p hairpin inhibitor, and their controls with unrelated sequences (Dharmacon, Lafayette, CO) were transfected at 50 nmol/l. Transfections were performed by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), as described by the manufacturer. After 6 hours, the transfection mix was replaced with complete medium. The analyses were performed 72 hours after transfection.
Generation of mimic miR-423-5p HCC cell lines using lentiviral transduction system
HCC cell lines HepG2, Hep3B and SNU387 were seeded in a 96 multiwell plate and the following day infected with empty vector (MISSION® pLKO.1-puro Empty Vector Control Plasmid DNA; Sigma-Aldrich) or miRIDIAN microRNA hsa-miR-423-5p shMIMIC lentiviral particles (Lentiviral Human TurboGFP shMIMIC; Thermo Scientific) at a multiplicity of infection (MOI) between 2.5 and 20. At 48 h post-infection, GFP fluorescent cells were detected by fluorescent microscopy and Puromycin (Invitrogen) was added to the medium (1.5 \(\mu\)g/ml and 2.5 \(\mu\)g/ml - established by killing curve assay performed on the wild type cells). The medium was changed every 2 days, and puromycin-resistant cells were selected and expanded. Successful transduction was assessed by positive tGFP expression, and miR-423-5p expression was confirmed by analyzing mature miR-423-5p expression using RT-qPCR. Hep3B LUC cells were obtained with a stable infection with lentiviral luciferase vector pRRLSIN.cPPTLuciferase (Addgene, Watertown, Massachusetts, USA).
Bacteria transformation and plasmids amplification
The pMD2.G, VSVG (Envelope), psPAX2 (Packaging), pLKO.1-puro Empty plasmids were amplified individually using XL1-Blue super-competent bacteria. First, the cells were thawed on ice for 5 minutes and mixed before transferring 50 \(\mu\)L to a chilled sterile polypropylene culture tube. 100 μg of each plasmid were added to the competent cells and placed on ice for 10 minutes. Cells were heat-shocked at 42 °C for 45 seconds, placed on ice for 2 minutes, added to 450 \(\mu\)L of lysogeny broth (LB; Sigma-Aldrich) containing 100\(\mu\)g/mL ampicillin (Sigma-Aldrich) and were shaken at 37 C° for 60 minutes. All 500 \(\mu\)L aliquot was plated on ampicillin-containing LB-Agar plates overnight, and afterwards, clones were picked and shaken overnight at 37 °C in 5 mL of LB broth containing ampicillin. The plasmid was extracted using the QIAprep® Miniprep (QIAGEN). Cells were pelleted in a centrifuge and resuspended in 250 \(\mu\)L of Buffer P1. 250 \(\mu\)L of Buffer P2 was added and mixed by inverting the tube. Then 350 \(\mu\)L of neutralization solution (N3) was added and mixed by inverting the tube. Cells were centrifuged for 10 minutes at 14,000 x g. The supernatant was transferred to a QIAprep 2.0 spin column and centrifuged for 1 minute at 12,000 x g. The flow-through was discarded, and two washes of 500 \(\mu\)L wash solution were performed, and the column was centrifuged for 1 minute at 12,000 x g each time. The plasmids were eluted from the column by adding 50 \(\mu\)L of elution buffer, waiting 2 minutes and centrifuging the columns at 12,000 g for 2 minutes. The concentration of the plasmids were then measured using a NanoDrop ND1000.
Lentiviral particles production
Target sequence plasmid, together with appropriate packaging plasmid and envelope plasmid, were transfected in HEK293T (Passage 3 maximum) using lipofectamine 2000. After 24 hours from transfection, HEK293T media containing Lentiviral Particles Fraction 1 was collected, filtered with a 0.22 μM syringe filter and split evenly in 1 mL cryovials for storage at −80°C. Fresh media was added to cells. After 48 hours from transfection, HEK293T media containing Lentiviral Particles Fraction 2 was collected, filtered with a 0.22 μM syringe filter, and split evenly into 1 mL cryovials for storage at −80°C. 1 mL of LVPs was able to transduce efficiently 80% of Wild Type cells (measured by GFP fluorescence) seeded in 6 multiwell plate.
Generation of MALAT-1 HCC cell lines using a lentiviral transduction system
HCC cell lines Hep3B and SNU387 were seeded in 6 multiwell plates and treated with 1 mL MALAT-1 lentiviral particles after 24 hours (HEK293T cell product, LVP titer not assessed). At 48 h post-infection, GFP fluorescent cells were detected by fluorescent microscopy and Puromycin (Invitrogen) was added to the medium (1.5 \(\mu\)g/ml established by Killing curve assay). The medium was changed every 2 days, and puromycin-resistant cells were selected and expanded. Successful transduction was assessed by positive tGFP expression, and MALAT-1 expression was confirmed by analysis of the long non-coding RNA expression using RT-qPCR.
RNA immunoprecipitation
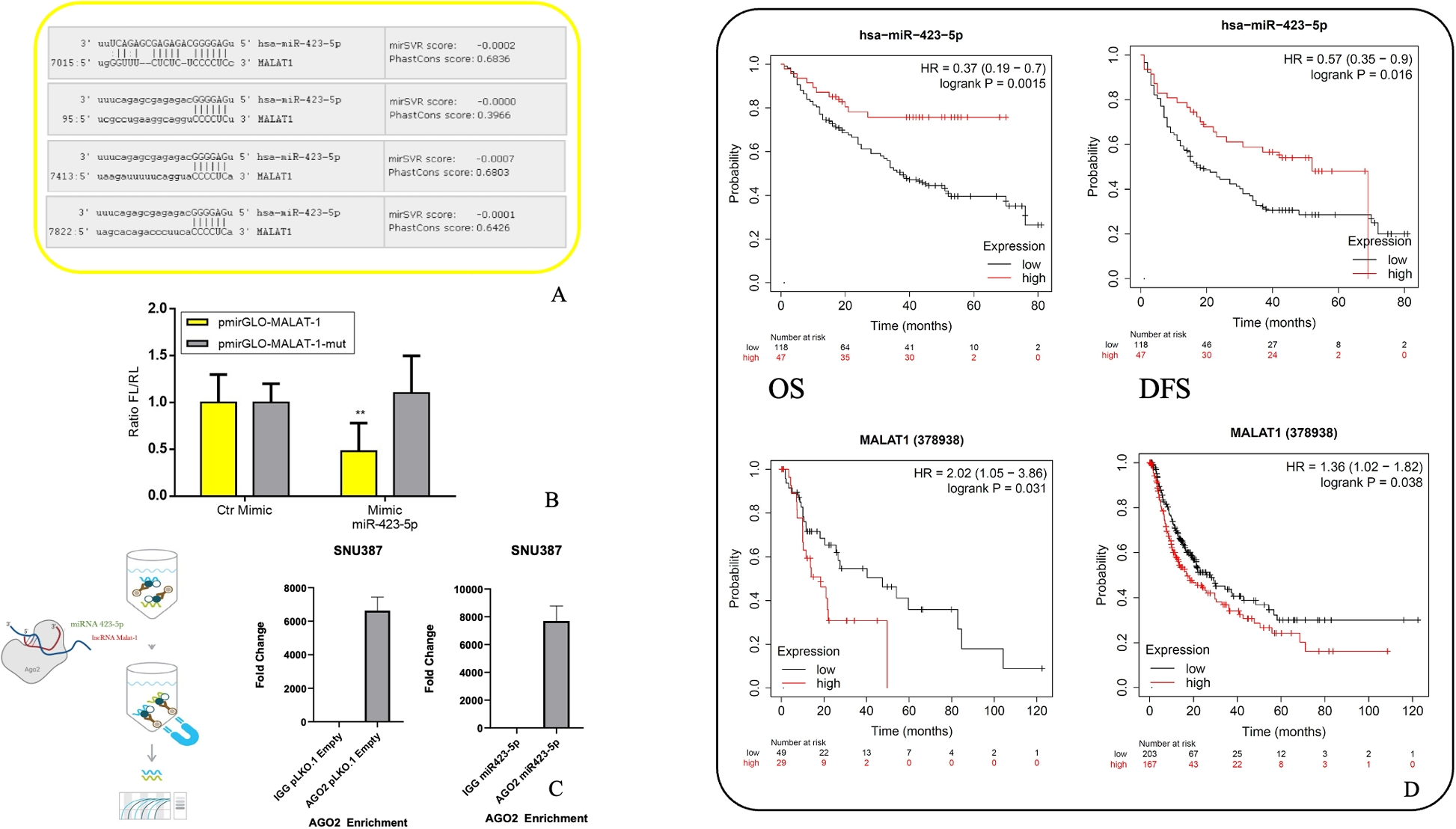
The RNA immunoprecipitation (RIP) assay was performed using extract obtained from SNU387 clones with Magna RIP RNA-binding protein immunoprecipitation kit (MilliporeSigma) according to manufacturer protocol. Briefly, the cells were scraper detached and lysed, the magnetic beads were prepared using AGO2 RIP antibody (MilliporeSigma) and a negative control IgG antibody. Immunoprecipitation was conducted overnight at 4 °C, then RNA purified with phenol-chloroform-isoamyl alcohol. FOS primers were used to check AGO2 enrichment and successful specific immunoprecipitation by qPCR while target RNAs related cDNA and expression analysis by qPCR was performed according to the methods described below.
CyQuant proliferation assay
Cells were detached from respective plates and seeded in a 96-well plate at a density of 1500 per well in sextuplicate for the CyQUANT NF Cell Proliferation Assay (C35006, Thermo Fisher Scientific). At 24 and 48 hours after seeding, cells were treated with the CyQUANT reagent for 30 minutes. The fluorescence readout was measured using the plate reader Infinite M200 Pro TECAN (Tecan Group Ltd, Männedorf, Switzerland) at 485 nm excitation and 530 nm emission. The CyQUANT NF Assay is based on the measurement of cellular DNA content via fluorescent dye binding. Because cellular DNA content is highly regulated within a specific cell population, it is closely proportional to cell number. All proliferation experiments were performed at least three times independently.
Proliferation assay – live-time imaging with IncuCyte
Cells were detached from respective plates and seeded in a 96-well plate at a density of 2500 and 5000 per well in sextuplicate for each cell concentration. Plates were then stored into IncuCyte Live-Time microscope incubator (37°C, 5% CO2) for a maximum of 120 hours. Capture protocol was set for 3 different captures per well, once every 30 minutes. The confluence percentage was recorded and plotted by IncuCyte software considering all the captures for each time point.
Cell migration and invasion assays
For wound healing assay (Scratch assay), HCC cells were cultured to 80% confluence and serum-starved for 24 h in 24 multiwell plate, then vertical scratches were drawn through the confluent monolayer of cells using a 10-\(\mu\)l pipette tip. After being washed with PBS to remove any cell debris caused by induction of the wound, images from triplicate experiments at 0 h, 12 h, 24 h were taken for Hep3B, and at 0 h, 24 h, 48 h were taken for HepG2 and SNU387. The distances between the scratch edges were measured at three different points using Carl Zeiss AxioVision software (Carl Zeiss, Oberkochen, Germany). The measurements were expressed as percentages of the wound area. Independent experiments were performed three times, with three separate wells per condition. For the chamber-based cell migration and invasion assay Cultrex 96-Well BME Cell Invasion Assay (3455-096-K, Cultrex) was used following the manufacturer’s recommendations (Trevigen, Gaithersburg, MD, USA). These assays employ a simplified Boyden chamber design with an 8-micron polyethylene terephthalate (PET) membrane. Briefly, the top chamber was coated with 0.5 X BME solution at 37 °C in a CO2 incubator for 4 hours and then added cells suspension in serum-free medium. The bottom chamber was filled with FBS containing medium and incubated at 37 °C with 5% CO2 for 48 hours. After washing the cells in the bottom chamber, they were labelled by Calcein AM (Trevigen), and the measure of the number of cells invaded or migrated was performed using the plate reader Infinite M200 Pro TECAN (Tecan Group Ltd, Männedorf, Switzerland) at 485 nm excitation and 520 nm emission. All experiments were independently repeated at least three times.
Colony formation assay
HCC cell lines were seeded in 6 MW at low density (2.5 × 105 cells/well) in culture media with low percentage FBS (1%). After 14 days, cells were stained with Crystal Violet, and the colonies were evidenced. The number and size of self-generated colonies were recorded and compared with the controls.
RNA extraction
Total RNA was extracted using the QIAGEN miRNeasy mini kit (QIAGEN, Cat-74104) or NORGEN Total RNA Purification Kit (Cat. 17200, 37500, 17250) following the manufacturer’s instructions. Briefly, pellet cells were lysed directly adding 250 \(\mu\)L Buffer RTL + 2 \(\mu\)L beta-mercaptoethanol. Then, 1 volume of 70% ethanol was added to the lysate, mixed well and transferred in a Mini spin column for centrifuge at 8000 x g for 15 sec. The columns were washed once with 350 \(\mu\)l buffer RW1 and twice with 500 \(\mu\)l buffer RPE with centrifugation at 8000 g for 1 min at room temperature between each wash. The columns were placed into a fresh collection tube and centrifuged at 14000 g for 2 min at room temperature to remove any buffer contaminants. Spin columns were then transferred to new RNase-free tubes and RNA was eluted by adding 50 \(\mu\)l RNase-free H2O to the columns and centrifuging at 8000 g for 1 min at room temperature. RNA was stored at −20°C for a short time. Total RNA was quantified using the NanoDrop 8000 Spectrophotometer (Thermo Scientific).
miRNA reverse transcription for miRNA expression analysis
For miRNA expression detection, cDNA was synthesized using stem-loop reverse transcription primers using the TaqManTM microRNA reverse transcription kit (Applied Biosystems, Paisley, UK) according to the manufacturer’s instructions. Each reaction contained 5 \(\mu\)l of 2 ng/\(\mu\)l sample RNA, 0.15 \(\mu\)L of 100mM dNTPs, 1\(\mu\)L 50U/\(\mu\)l MultiScribe Reverse Transcriptase, 0,19 \(\mu\)L 20U/\(\mu\)l RNase Inhibitor, 1,5 \(\mu\)L 10 X Reverse Transcriptase buffer, 4.16 \(\mu\)l RNase-free water and 3\(\mu\)L Taqman® miRNA Reverse Transcription Primers 5X for the mature sequence of the miRNA of interest. Endogenous control RNU6 was also performed for each sample to normalize changes in the miRNA expression. The mature sequence of miRNA of interest and endogenous control sequences are: - hsa-miR-423-5p 5’ UGAGGGGCAGAGAGCGAGACUUU 3’ - U6snRNA 5’GTGCTCGCTTCGGCAGCACATATACTAAAATTGGAACGATACAGAGAAGATTAGCATGGCCCCTGCGCAAGGATGACACGCAAATTCGTGAAGCGTTCCATATTTT 3’. cDNA synthesis reactions were incubated at 16 ºC for 30 min, 42 ºC for 30 min, and 85 ºC for 5 min and then held at 4 ºC. Samples were stored at −20 ºC before quantitative PCR was performed.
mRNA reverse transcription for gene expression analysis
For first-strand cDNA synthesis from RNA was prepared following the manufacturer’s instructions described below. Into a 0.5ml Eppendorf tube, 2 \(\mu\)g of RNA, 1 \(\mu\)l Oligo (DT)15 primer (Promega) and ddH2O were added to achieve a final volume of 10 \(\mu\)l. The samples were then heated using UNO-Thermoblock to 70 °C for 5 minutes to melt secondary structures. Samples were then immediately put on ice for 5 minutes before the following were added to each sample: 5 \(\mu\)L M-MLV 5X Reaction Buffer (Promega), 1\(\mu\)L M-MLV Reverse Transcriptase (Promega), 0.7\(\mu\)L RNasin Ribonuclease Inhibitor (Promega), 1\(\mu\)L Deoxynucleotide Set (dNTPS) (Sigma) and 7.3 \(\mu\)L Nuclease Free water. Samples were mixed gently and incubated for 60 minutes at 40 °C in a water bath. After incubation, samples were finally heated using an UNO-Thermoblock to 95 °C for 5 minutes and immediately stored at −20°C, ready for qRT-PCR.
TaqMan quantitative real-time PCR
miRNA qRT-PCR was performed using Taqman® miRNA Assay RT-PCR Probes and TaqMan® Universal Mastermix PCR (containing AmpliTaq Gold® DNA polymerase, dNTP mixture and optimal salt conditions, Invitrogen) on Rotor-Gene PCR cycler (Qiagen) in accordance with the manufacturer’s instructions. Each reaction master mix contained 250 nM TaqMan probe, 1 X Taqman Universal PCR MasterMix II (Invitrogen) and nuclease-free H2O to a total volume of 18,67 \(\mu\)l per sample. cDNA was pipetted in technical triplicate, 1.33 \(\mu\)l per well, and the master mix was added to each well to give a total reaction volume of 20 \(\mu\)l. RNU6 was used as an endogenous control in separate wells. Thermal cycling as follows: 2 min incubation at 50ºC, 10 min incubation at 95 ºC then 40 cycles of 15 sec at 95 ºC and then 60 sec at 60 ºC.
Quantitative qRT-PCR for mRNA expression
qRT-PCR was set up into PCR tubes using Sybr Green SuperMix on Rotor-Gene PCR cycler (Qiagen) following manufacturer’s instructions described here per sample: 6.75 \(\mu\)L iTaq™ Universal SYBR® Green Supermix (Bio-Rad), 0.5 \(\mu\)L 10 pM Forward Primer (MALAT-1 5’ GGATCCTAGACCAGCATGCC 3’; GAPDH 5’ ACCCACTCCTCCACCTTTGA 3’ HPRT1 5’ CATTATGCTGAGGATTTGGAAAGG 3’), 0.5 \(\mu\)L 10 pM Reverse Primer (MALAT-1 5’AAAGGTTACCATAAGTAAGTTCCAGAAAA 3’; GAPDH 5’ CTGTTGCTGTAGCCAAATTCGT 3’ HPRT1 5’ CTTGAGCACACAGAGGGCTACA 3’) and 2.25\(\mu\)L Nuclease Free water for a total volume of 10 \(\mu\)L. 2 \(\mu\)L of 50 ng cDNA was pipetted in technical triplicate per well and the master mix added to each well to give a total reaction volume of 12 \(\mu\)l. GAPDH was used as an endogenous control in separate wells. In cases of Non-Template Controls (NTC), cDNA template was substituted with ddH2O. Thermal cycling conditions began with 5 min incubation at 95 °C, then followed by 40 cycles of 10 sec at 95 ºC, 20 sec at 60 ºC and then 20 sec at 72 ºC. The primers used are design and product by Eurofins Genomics. Additional Melt analysis to assess the quality of the experiment is performed at the end of the run. Mitochondrial related transcripts primers are reported in Supplementary Table 2. Results were shown relative to the control sample using the \(\Delta\)Ct method and expressed as relative fold change. For miRNA expression, results were normalised to U6. For mRNA and long non-coding RNA expression, results were normalised to GAPDH or HPRT1.
Fluorescent mitochondria cell lines generation
HCC cell lines were transfected with MTS-mCherry-GFP1-10-Hyg-N1 (Addgene plasmid # 91957) using Lipofectamine 2000 according to the manufacturer protocol. Pure population was then selected using G418 antibiotic (Sigma-Aldrich). Mitochondria were visible (Red) in vital cells using fluorescent microscopy (LEICA DM 6000 B 100x and 60x Oil), and the number and shape were recorded and compared between the cell lines.
Transcriptome analysis
RNA was extracted from HCC cell clones using Total RNA Purification Kit (Norgen Biotek Corp) according to the manufacturer protocol. Each cell clone was cultured in three separate flasks to achieve biological triplicates. RNA quantification and quality assessment were performed using NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific), Qubit RNA HS Assay Kit (Thermo Fisher Scientific) and Bioanalyzer Total RNA Pico Chip (Agilent). SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific) was used to obtain cDNA according to manufacturer protocol. IonChef (Thermo Fisher Scientific), related Ion AmpliSeq Kit for Chef DL8 (Thermo Fisher Scientific) and Trascriptomic panel (Thermo Fisher Scientific) were used to obtain sequencing libraries and sequencing chip template. The NGS runs were performed on IonS5 (Thermo Fisher Scientific) on Ion 540 Chip (Thermo Fisher Scientific).
In vivo experiment
All animal procedures were in compliance with the national and international directives (D.L. March 4, 2014, no. 26; directive 2010/63/EU of the European Parliament and European Council; Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 2011;Animal Research guidelines Reporting of In Vivo Experiments (ARRIVE) guidelines) and approved by the Italian Ministry of Health (authorization n.346/2020-PR, released on April 2020). Mice were maintained in a barrier facility on high-efficiency particulate air HEPA-filtered racks and received food and water ad libitum. NOD SCID (6 weeks old) male immunodeficient mice (Charles River Laboratories, Calco, Italy) were orthotopically injected in the liver with 1\(\times\)106 of Hep3B control or miR-423-5p-LUC over expressing cells using an insulin syringe with a 27-gauge needle. Mice were anesthetized with a combination of tiletamine–zolazepam (Telazol, Virbac, Carros, France) and xylazine (xylazine/Rompun, Bayer, Leverkusen, North Rhine-Westphalia, Germany) given intramuscularly at 2 mg/kg. Following the anesthesia, a parallel incision to the linea alba is made in the abdominal wall to expose the liver. Then, 10 μl of Hep3B cells are mixed with 10 μl of Matrigel and injected in the liver very slowly. After injection, the peritoneum was sealed using absorbable suture (PolySorbTM 6-0) and the skin layer of the abdominal wall was close by absorbable suture (PolySorbTM 5-0). Finally, mice were medicated with an oral administration of 0.5 mg/Kg of Metacam (meloxicam) to control post-operative pain and inflammation. Real time tumor growth was monitored using the IVIS Lumina II CCD camera system (PerkinElmer, Shelton, Connecticut, USA) by intraperitoneal injection with 150 mg/Kg D-Luciferin (PerkinElmer). Bioluminescence signals were determined by the number of photons and were acquired and analyzed using the Living image software version 4.3 (PerkinElmer). Each experimental group included 5 mice. For the antisense GapmeR-MALAT-1 experiment, NOD SCID (6 weeks old) male immunodeficient mice were orthotopically injected in the liver with 1\(\times\)106 Hep3B-LUC cells as described above. Seven days after cell injection, mice were randomized and treated intraperitoneally (ip) with antisense LNA negative control (Qiagen n.3545711) at 25 mg/Kg twice a week for five total treatments, with antisense LNA GapmeR-MALAT-1 (Qiagen n.3545711) at 25 mg/Kg twice a week. Real time tumor growth was monitored weekly using the IVIS Lumina II CCD camera system (PerkinElmer, Shelton, Connecticut, USA) by intraperitoneal injection with 150 mg/Kg D-Luciferin (PerkinElmer). Bioluminescence signals were determined by the number of photons and were acquired and analyzed using the Living image software version 4.3 (PerkinElmer). Each experimental group included 6 mice. The student’s t-test (unpaired, two-tailed) was used for single pair-wise comparisons. Differences were considered statistically significant when \(P < 0.05\). The student’s t-test (unpaired, two-tailed) was used for single pair-wise comparisons. Differences were considered statistically significant when \(P < 0.05\).
Statistical and bioinformatic analysis
All data are presented as the mean ± SEM for at least three independent experiments. For each experiment, the statistical tests are indicated in the results section. The student’s t-test was conducted using Prism 8 (Graphpad Software, La Jolla, CA, USA) to compare the means. Significant (‘*’/\(p= < 0.05\)), very significant (‘**’/\(p= < 0.01\)), highly significant (‘***’/\(p= < 0.001\)) or very highly significant (‘****’/\(p= < 0.0001\)). Other parametrical and non-parametrical analyses were carried out using IBM SPSS Ver. 25, p-value \(<0.05\) is considered statistically significant. Survival analysis and Kaplan-Meier curves for Overall Survival (OS) and Disease-Free Survival (DFS) were generated using the Kaplan-Meier Plotter online tool (KM Plotter, http://kmplot.com/analysis), which integrates publicly available transcriptomic datasets from liver cancer patients. Differential expression analyses were performed using DESeq2 v1.36.0 R package (1). Before performing the differential expression analyses the genes with 10 counts in at least two samples were selected. Genes with an absolute value of log2 fold-change (FC) greater than 1 and adjusted p-value less than 0.01 were selected as differentially expressed for each comparison. The up- and down-regulated genes selected for each comparison were used to perform a gene set enrichment analysis with DAVID web server (2, 3). Biological processes with Fisher-exact p-value less than 0.05 were selected for downstream analyses. The circos plots were made using the circlize R package (4). The analyses were performed using R Statistical Software v4.2.1.





Comments (0)