Patient samples
Human cholangiocarcinoma tissue samples were obtained from the Department of Biliary and Pancreatic Surgery at Tongji Hospital, Huazhong University of Science and Technology (Wuhan, China). These samples were surgically resected and confirmed as cholangiocarcinoma through pathological examination. Patients underwent postoperative follow-up until either their date of death or the last recorded follow-up. The collection procedures for all tissue samples were approved by the Ethics Committee of Tongji Hospital, Huazhong University of Science and Technology (HUST), in accordance with the principles outlined in the Declaration of Helsinki.
Cell culture
Human cholangiocarcinoma cells (TFK1, HuCCT1) and human embryonic kidney cells (HEK293T) were cultured in RPMI 1640 or high-glucose DMEM medium (Servicebio, China) supplemented with 10% fetal bovine serum (NEWZERUM, New Zealand) at 37 °C and 5% carbon dioxide. All the cell lines were subjected to Mycoplasma infection testing and confirmed to be free of any infections.
Plasmid construction
The full-length coding sequences (CDSs) of STUB1, UHRF1, and PLA2G2A were amplified from human cDNA via PCR and subsequently cloned and inserted into the pHAGE vector. Truncated fragments of UHRF1 and STUB1 were amplified via PCR via UHRF1-Flag and STUB1-Flag plasmids, respectively. Gene-specific small hairpin RNA primers were provided by Sangon Biotech Co., Ltd., and inserted into the pLKO.1 vector. The primer sequence information can be found in (Additional file 1).
Viral packaging and viral infection
The target gene plasmid was cotransfected with the pMD2G and psPAX packaging plasmids into HEK293T cells. After 72 h of transient transfection using Polyethylene Linear (PEI) MW40000 (40816ES02, Yeasen, China), the supernatant was collected and filtered through a 0.45 μm filter (BS-PES-45, Biosharp, China). TFK1 and HuCCT1 cells were cultured in 1 mL of virus supernatant and 1 mL of RPMI 1640 complete medium supplemented with 2 µL of polybrene (40804ES76, Yeasen, China). After 24 h of transfection, puromycin hydrochloride (CL13900, Selleck, USA) was used for screening at a concentration of 1 µg/mL for 2 weeks. Then, Western blot experiments were conducted to assess the knockdown or overexpression efficiency of the target genes.
Cell proliferation assay
A Cell Counting Kit-8 (CCK-8) (40203ES92, Yeasen, China) was used to assess cell viability. A total of 1000 stably transfected cells were seeded in 96-well plates and treated with the CCK-8 reagent working solution at different time points. The cells were incubated at 37 °C and 5% CO2 for 90 min, after which the absorbance was measured at a wavelength of 450 nm. For the colony formation experiment, 1000 cells were plated in 6-well plates with 2 mL of fresh culture medium and allowed to grow for 2 weeks. Staining was subsequently performed using a solution of 1% crystal violet to calculate the number of cell clones.
Wound healing assay
The cells were evenly distributed in a 6-well plate, and scratches were induced using a 200 µL pipette tip after cell apposition. The scratched cells were subsequently removed by washing with PBS. Images were captured at 0 and 72 h after scratching, and the area of cell migration was quantified using ImageJ software.
Transwell assay
To perform the invasion assay, we precoated Transwells with 100 mL of Matrigel at 37 ℃ for 1 h. Subsequently, 200 µL of serum-free medium containing 3 × 104 cells was added to the center of each Transwell (polycarbonate membrane with a pore diameter of 8 μm; Corning Incorporated). The insert was then placed in a container filled with 800 µL of cell culture medium supplemented with 10% fetal bovine serum as the receiving well. Following incubation for either 48–72 h, the inserts were retrieved and stained with a solution of 1% crystal violet.
Co-immunoprecipitation
The cells were transfected with the corresponding plasmids for 24 h, followed by cell lysis using IP buffer and the addition of complete protease inhibitors (REF: 04693132001, Roche, Indianapolis, IN). Ultrasound and centrifugation were performed on the cell lysate. A total of 40 µL of the supernatant from the cell lysis solution was used as the input, and the remaining portion was incubated with A/G magnetic beads (B26102, Biomake, USA) and the corresponding antibodies at 4 °C for 12 h. Subsequently, 5 rounds of washing of the magnetic beads with precooled IP buffer were performed for 5 min each. Finally, 40 µL of loading buffer was added to the magnetic beads, which were boiled at 95 °C for 15 min. The mixture was subsequently centrifuged, and the supernatant was collected for immunoblotting and subsequent analysis. IgG was used as a control for immunoprecipitation. The antibodies used are listed in (Additional file 2). The UHRF1 mass spectrometry binding protein obtained through co-immunoprecipitation is presented in (Additional file 3).
GST pull-down assay
The CDS region of STUB1/UHRF1 was amplified via PCR and subsequently cloned and inserted into the pGEX-4T-1 vector. Escherichia coli BL21 were transformed with the resulting plasmid. Following the addition of IPTG (final concentration of 500 µM), bacterial cultivation was carried out at 16 °C for 12 h. Subsequently, the bacteria were lysed using GST buffer, followed by sonication and centrifugation to collect the supernatant. The supernatant was then incubated with GST magnetic beads on a rotating device at 4 °C for 1 h. Then, 5 washes with GST buffer for 5 min each were performed to remove nonspecific binding. The obtained GST magnetic beads were mixed with the cell lysate and incubated on a rotary shaker at 4 °C for 4 h. Following this step, the magnetic beads were washed 5 times with precooled IP buffer for 5 min each time. Finally, the magnetic beads were boiled in loading buffer at 95 ℃ for 15 min to elute bound proteins from the beads. The supernatant was collected after centrifugation and subjected to immunoblotting analysis.
Nuclear and cytoplasmic protein purification and isolation
A Nuclear Protein and Cytoplasmic Protein Extraction Kit (P0028, Beyotime) was used to isolate the nuclear and cytoplasmic proteins. The cells were subsequently washed with PBS, and the cytoplasmic proteins were extracted using cytoplasmic protein extraction reagent A/B, whereas the nuclear proteins were extracted using nuclear protein extraction reagent. The proteins were subsequently subjected to analysis via SDS‒PAGE and immunoblotting.
Western blot
Western blot analysis was performed by adding RIPA buffer supplemented with a cocktail of protease inhibitors to the lysed cell samples. Total protein extracts were mixed with loading buffer and denatured at 95 ℃ for 15 min. The proteins were subsequently separated via SDS‒PAGE and transferred onto a PVDF fiber membrane. The membrane was then blocked with 5% BSA at room temperature and incubated with primary antibodies at 4 ℃. Next, the membrane was incubated with the secondary antibody, and signal visualization was achieved via enhanced chemiluminescence.
Total RNA isolation and RT‒qPCR
The cells were processed according to the experimental requirements. Total RNA was extracted using TRIzol (Vazyme, Nanjing, China), followed by reverse transcription of the RNA using HiScript®III RT SuperMix for qPCR (Vazyme). A quantitative analysis was subsequently performed using a PCR instrument that combines cDNA, SYBR Green dye (Vazyme), and the specific primers listed in (Additional file 4).
Bisulfite sequencing PCR (BSP)
BSP primers were designed according to the online MethPrimer program (http://www.urogene.org/methprimer). The primers used can be found in (Additional file 5). DNA extraction was performed using a DNA extraction kit (D3396020000J12T006, Omega, USA), and bisulfite conversion for the BSP conversion reaction was carried out using a kit (EM101-02, Vazyme, China). PCR amplification was subsequently conducted using the Taq enzyme (C601-02, Vazyme, China). After purification of the PCR products, connection with the vector (EM101-02, Vazyme, China) was performed, and 10 bacterial clones were randomly selected for sequencing.
Immunofluorescence staining
TFK1 and HuCCT1 cells were seeded on glass slides in 12-well plates. After 48 h, the cells were fixed with a 4% paraformaldehyde solution for 15 min and permeabilized with 0.1% Triton X-100 for 10 min. Subsequently, the cells were incubated overnight at 4 ℃ with primary antibodies, followed by incubation with secondary antibodies at room temperature for 1 h. Finally, cellular observations and imaging were performed using laser confocal microscopy. ImageJ software was used for further relative quantitative analysis, with the DAPI staining mask employed to define the regions of interest (ROIs) within the nucleus, thereby effectively distinguishing it from the cytoplasm. To eliminate artificial differences in staining intensity, we compared the staining intensities of the cytoplasm and nucleus to determine the cytoplasmic: nuclear ratio as a relative measure of UHRF1 nuclear localization [36]. The 3D Surface Plot plugin in ImageJ was used to visualize the relative fluorescence intensity of UHRF1 in the cytoplasm, where the luminance of the image was interpreted as the height of the plot [37]. The specific method can be accessed in the user guide for ImageJ (https://imagej.net/ij/plugins/surface-plot-3d.html).
Experiments with animals
BALB/c (nu/nu) female nude mice and male C57BL/6J mice were procured from Jiangsu Jicui Yaokang Technology Co., Ltd. and housed in a specific pathogen-free facility at the Animal Center of Tongji Hospital, affiliated with Tongji Medical College, Huazhong University of Science and Technology.
For the xenograft experiments, 5-week-old female BALB/c (nu/nu) nude mice were randomly divided into groups (n = 8 in each group). A total of 3 × 106 stably transduced cells were resuspended in 100 µL of PBS and injected into the dorsal side of each mouse. The experimental mice were euthanized by cervical dislocation, and the tumors were subsequently excised for further analysis.
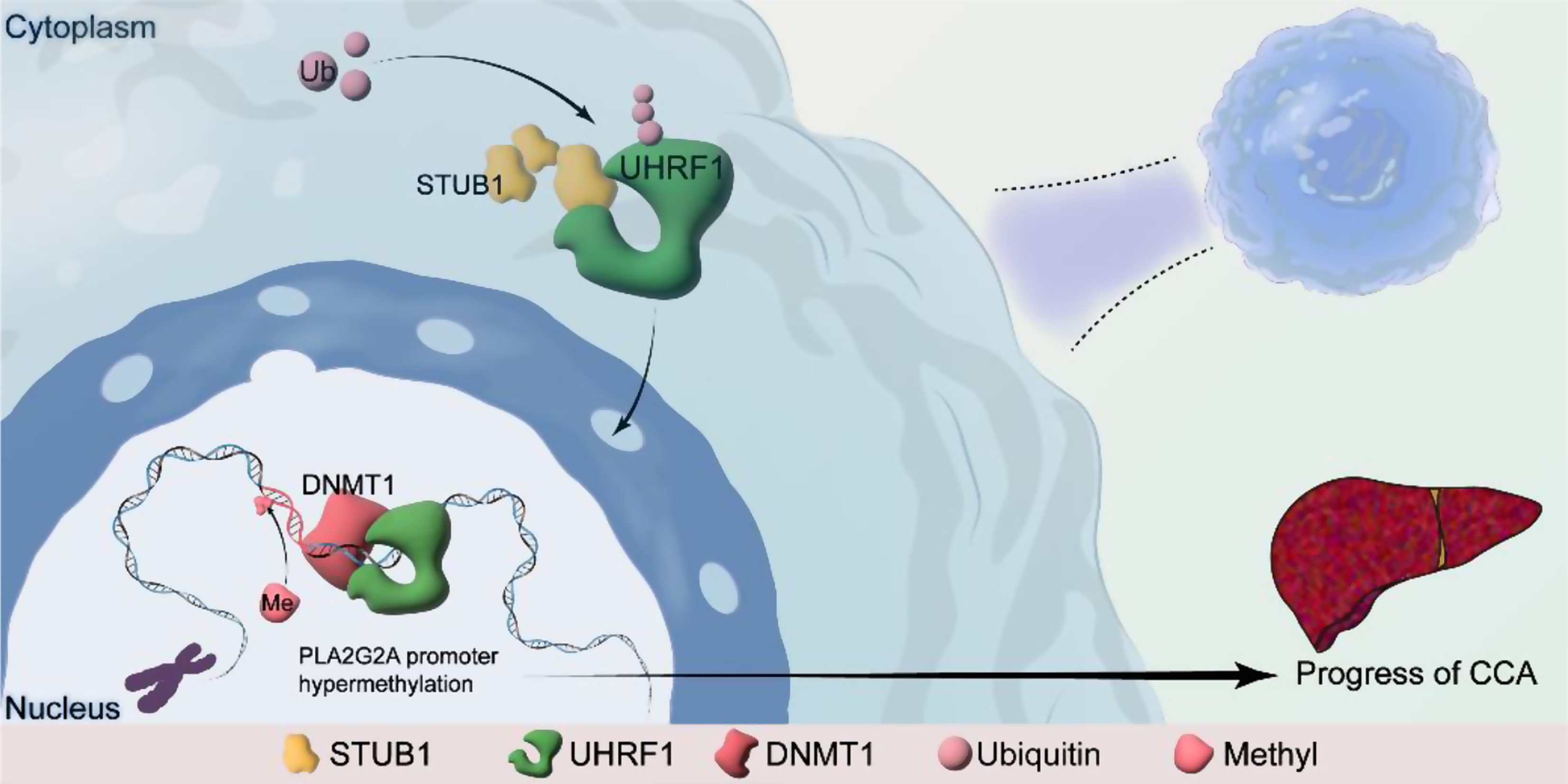
To establish a mouse model of primary cholangiocarcinoma, we diluted 20 µg of pT3EF1aH-myr-Akt (179909, Addgene, USA) and 20 µg of pT3EF1aH-NICD1 (86500, Addgene, USA), along with 6 µg of the transposable plasmid pCMV (CAT) T7-SB100 (34879, Addgene, USA), in 2 mL of physiological saline. Subsequently, 2 mL from the plasmid mixture was injected into the lateral tail vein of male C57BL/6J mice (5 weeks old) within a time frame not exceeding 7 s. To overexpress the Uhrf1 and Stub1 proteins, we constructed the pT3EF1aH-Uhrf1/pT3EF1aH-Stub1 plasmid using the pT3EF1aH vector. An additional 20 µg of this construct plasmid was added to the aforementioned plasmid mixture. Liver samples were collected after hydrodynamic transfection for analysis of the occurrence and progression of CCA tumors.
Immunohistochemical (IHC) staining
The detailed experimental procedures and protocols can be found in our previous research [38].
Statistical analysis
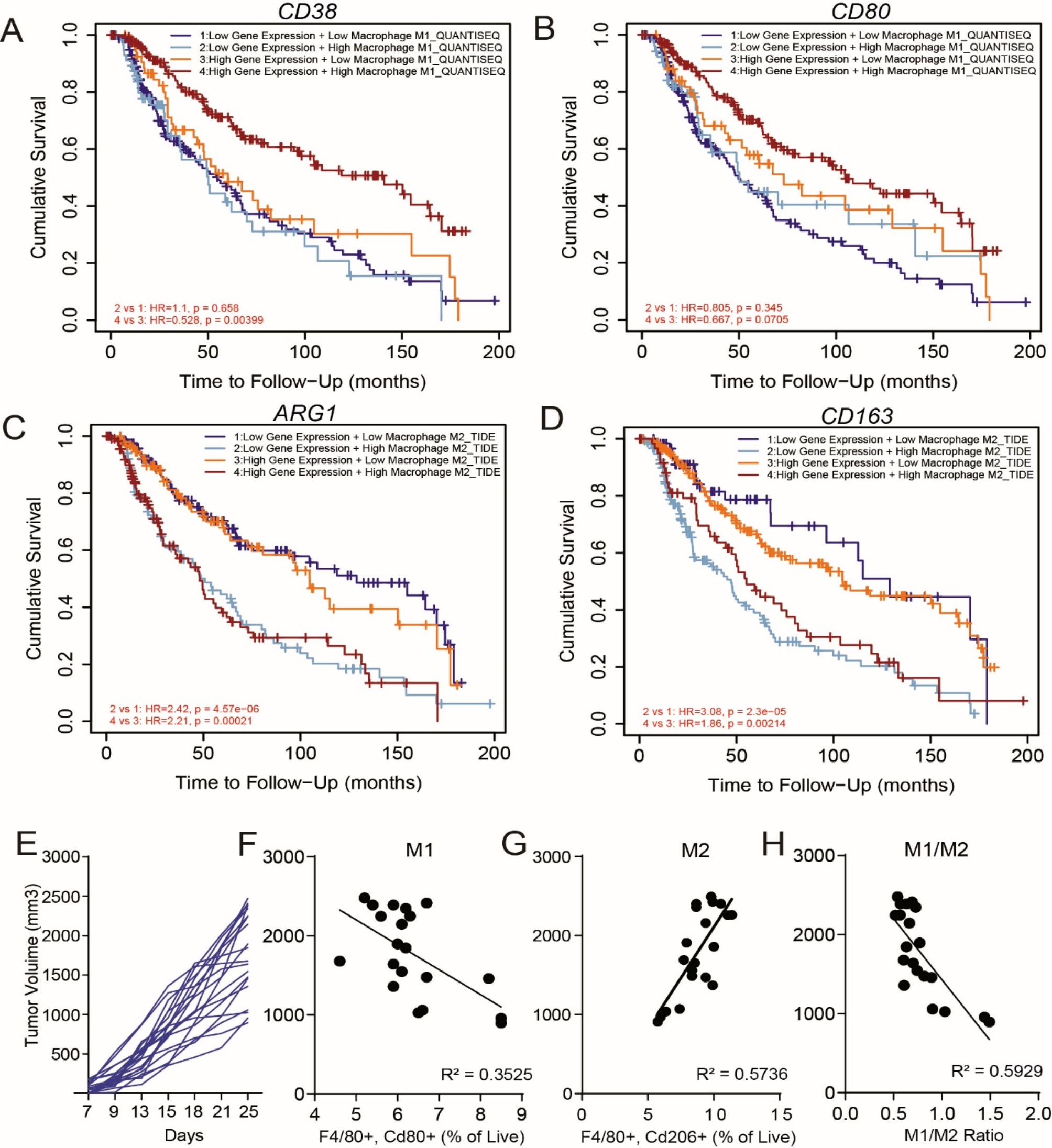
The experiments were conducted independently and repeated at least three times. The data are presented as the means ± standard deviations unless otherwise specified. Statistical analysis was performed using GraphPad Prism Software 9.0, a widely recognized tool in scientific research. Student’s t test was used to compare two independent groups. Survival curve analysis was performed using the Kaplan‒Meier method and log rank test. The images were processed and integrated via ImageJ-win64, Photoshop and Adobe Illustrator 2022 for display. A P value < 0.05 was considered statistically significant: * P < 0.05, ** P < 0.01, *** P < 0.001.





Comments (0)