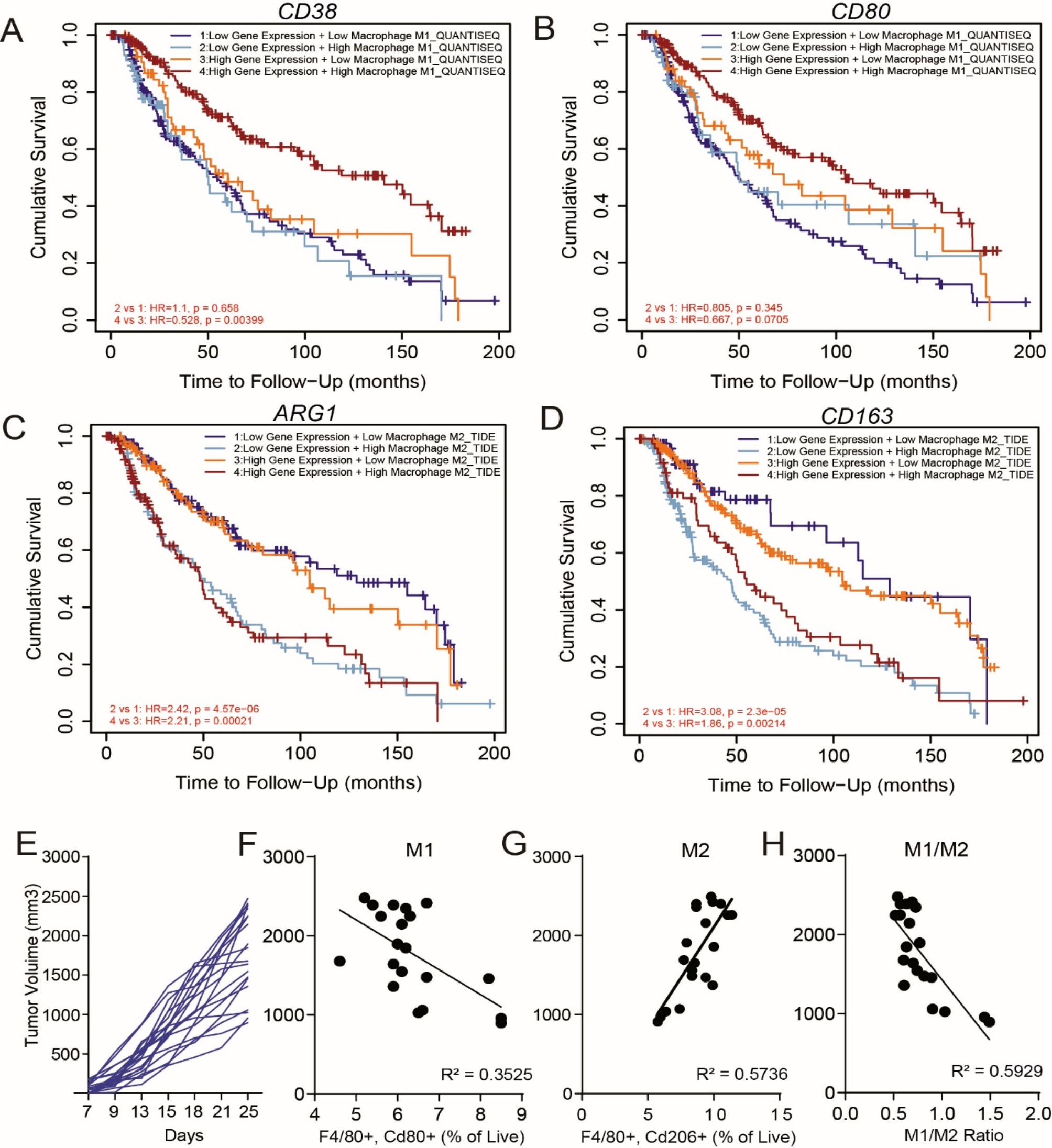
Cells and reagents
NRASmut human melanoma cell lines SKmel147 (Prof. Dr. Jochen Utikal, University Medical Center Mannheim, Germany), SKmel30 and MelJuso (DSMZ, Leibniz Institut, Germany), and the BRAFwt/NRASwt human melanoma cell line WM3918 (Rockland, USA) were cultured in RPMI 1640 enriched with GlutaMAX (Gibco Thermo Fisher Scientific, USA), supplemented with 10% FCS (Fetal Calf Serum, Gibco Thermo Fisher Scientific, USA) and 0.1 mg/mL Normocin (InvivoGen, USA). Patient-derived human melanoma cell lines M160915 and M161022 (Prof. Mitchell Levesque, University Hospital Zurich , Switzerland) were cultured in RPMI 1640 (Gibco Thermo Fisher Scientific, USA), supplemented with 10% FCS, 1mM Sodium Pyruvate (Gibco Thermo Fisher Scientific, USA), 4mM L-Glutamine (Gibco Thermo Fisher Scientific, USA), and 0.1 mg/mL Normocin. NHDF (normal human dermal fibroblasts) (Promocell, C-12300), MRC-5 (human lung fibroblasts) (ATCC, CCL-171), and LX-2 cells (human hepatic stellate cells) (Merk, SCC064) were cultured in DMEM enriched with GlutaMAX (Gibco Thermo Fisher Scientific, USA), supplemented with 10% FCS, 2.5% HEPES buffer 1 M (Gibco Thermo Fisher Scientific, USA), and 0.1 mg/mL Normocin. HMEC-1 (human endothelial cells) (ATCC, CRL-3243) were cultured in MCDB131 (Gibco Thermo Fisher Scientific, USA), supplemented with 10% FCS, 1 µg/mL Hydrocortisone (Sigma-Aldrich, USA), 10mM L-Glutamine, 0.1 mg/mL Normocin, and 10 ng/mL recombinant human EGF (PeproTech, USA). Trametinib (MEKi)-resistant SKmel30 and WM3918 cell lines were generated by continuous drug exposure of parental drug-sensitive cell lines to 5xIC50 and 1xIC50 concentrations, respectively, for approximately 3 months. The Binimetinib (MEKi)-resistant MelJuso cell line was generated by continuous drug exposure of the parental drug-sensitive cell line to 10xIC50 concentration of Binimetinib. All cell lines were transduced with Multiplicity of Infection (MOI) 3 of lentiviral vectors carrying reporter genes, for stable fluorescent protein expression. SKmel147, SKmel30, and M161022 were transduced with rLV.EF1.mCherry-9; NHDF, MRC-5 and LX-2 were transduced with pLenti-C-mGFP-P2A-Puro.; HMEC-1 were transduced with pLV-Bsd-CMV > tagBFP. After transduction, cells were subjected to antibiotic selection (either Puromycin or Blasticidin) and FACS-sorted using a BD FACSMelody™ Cell Sorter (BD Bioscences, USA). Cell growth was maintained at 37 °C in a humidified atmosphere comprising 5% CO2. All cell lines were regularly examined for mycoplasma contamination. Cell Line authentication was performed at Luxgen (Luxembourg).
The compound library Prestwick Chemical library® (PCL, Prestwick Chemicals, USA) is composed of 1267 mainly FDA-approved compounds supplied at 10mM concentration in DMSO. The in-house “Melanoma drug library” (MDL) was generated based on literature for their effect on the different melanoma genomic subtypes. It is composed of 61 compounds supplied at 10 mM concentration in DMSO, purchased from Selleckchem. Selected hit drugs were purchased individually from Prestwick Chemicals and dispensed in a specific ready-to-use source plate. For cell treatments outside the HTS workflow: Trametinib (#S2673), Daunorubicin HCl (#S3035), and Pyrvinium Pamoate (#S5816) were purchased from Selleckchem (Germany). Staurosporine (#CAYM81590-1) was purchased from Cayman Chemical (USA).
3D high-throughput screening
HTS assays were performed using the HTS platform, “Disease Modelling and Screening Platform” (DMSP) of LIH/LCSB, Luxembourg. The platform is equipped with two liquid handler workstations (Biomek NXp and Biomek FXp; Beckman Coulter), two integrated incubators (Cytomat 24-C; Thermofisher), an acoustic droplet ejector (Echo 550; Labcyte), a multimode plate reader (SpectraMax i3;Molecular Devices), a confocal high-content microscope (CV8000; Yokogawa) equipped with solid lasers (wavelengths: 405/488/561 nm) and emission filters (445/45 nm, 525/50 nm, 600/37 nm), and an integrated robotic arm on rail (SCARA; Beckman Coulter). Cells were seeded in 384-well U-bottom ULA black plates (Corning®, 4516, USA) at a density of 5 × 103 cells/well in 20 µL/well, centrifuged at 500 x g for 5 min, and incubated for 72 h at 37 °C and 5% CO2 to allow for spheroid formation. After the compounds from the PCL and MDL libraries were dispensed (one compound per well) at nanoliter range using the acoustic droplet ejector, a further 40 µL of fresh culture medium were added and spheroids were incubated for 5 days at 37 °C and 5% CO2. Every compound was dispensed at a final concentration of either 1 µM or 10 µM, each of them in duplicate (on two separate plates), with a final DMSO concentration of 0.1%. Side wells were dedicated to a pre-selected positive control compound for the screening (Foretinib 30 µM) and negative controls (DMSO 0.1%), and the first and last rows and columns of the plate were excluded to reduce edge effects. Additional plates were added to the screen with the compounds falling into the edge effect area. To detect and quantify the spheroid response to drugs we extracted the maximum intensity projection (MIP) area for each spheroid by applying high-content image analysis (see below). The MIPs were obtained from the Calcein AM (Cayman Chemical, USA) signal, thus representing a surrogate measure of cell viability, and informing on the size or growth of spheroids exposed to the drugs. At the end of the drug treatment, Calcein AM was added 4 X concentrated as 20 µL/well to reach a final concentration of 4 µM (80 µL final volume in each well) and incubated for 2 h at 37 °C. We initially used different Calcein AM concentrations and incubation times to optimize the ratio signal-to-noise to give us the most robust signal for imaging of spheroids. Confocal images were acquired using a 10x objective, 488 nm laser 525/50 nm emission filter, Z-stack acquisition (e.g. the Z-stack consisted of 40 slices taken sequentially with 10 μm step size for a total span of 390 μm) and on-the-fly generation of MIP images mode. A mock test was run before each HTS campaign to check the quality of the cells and assay, following the same seeding and timing procedures and including a drug response performed using a 3-fold dilution series of Foretinib starting at 10 µM.
Hit drug identification
CellPathFinder® was used to analyze MIP images and extract the total spheroid area in each well. In brief, a segmentation mask was created on the Calcein AM green-fluorescent signal, which allowed for the calculation of the radius and area of the spheroid MIP. The software summed all the area’s segments outputting a total Calcein AM area per well (µm2). The application of a statistical test (Grubb’s test), followed by visual inspection, removed outliers (such as failure of segmentation) from the set of data. The Z’-factor was calculated for each plate of the primary screening as a quality control step. The raw MIP measures were used to mathematically set a plate-specific cut-off for determining hit drugs, by applying the following formula: averageDMSO − (3 x standard_deviationDMSO) [14]. Drugs were taken into consideration if the raw MIP area values were below the cut-off in both duplicate plates. Data was normalized for the corresponding DMSO controls in the plate and expressed as percentage of residual MIP. Only drugs below 50% of this residual MIP in both duplicates from the set of values below the acceptable SD cut-off, were processed into the final selection step. Finally, we visually examined the MIP images to confirm the drug’s effect and rule out false positives. Additionally, we applied extra criteria, such as reviewing existing literature, to compile a final list of effective drugs. Rstudio was used for the analysis and the creation of relative plots.
Drug-response curve analysis in HTS fashion for hit validation
Drug-response curves (DRC) to determine the relative half-maximal inhibitory concentration (IC50) values were generated for the 17 selected hits using the same approach as described for the primary screening. Drugs, including the positive control Foretinib, were dispensed in duplicate using a 3-fold dilution series from the dedicated source plate, starting from 10µM with 10 dilutions. Cell viability was assessed using Calcein AM (as previously described). Data were normalized by the DMSO control within each plate. GraphPad 10.3.1 software (GraphPad, USA) software and non-linear regression (four parameters) analysis were used to extrapolate IC50 and R2 values for each tested compound.
3D mono- and multi-component spheroid generation
Mono-component spheroids were generated in 384-well ULA U-bottom plates (S-Bio®, MS-9384UZ, Japan) as follows: melanoma cells were seeded at a density of 0.5-1 × 103 cells/well in 80 µL of RPMI. The plate was centrifuged 500 x g for 5 min and incubated at 37 °C and 5% CO2 for 96 h.
Multi-component spheroids were generated as described before [11]. Melanoma cells, fibroblasts or hepatic stellate cells, and endothelial cells were seeded at a cellular ratio of 1:3:3 in 384-well black/clear round bottom ultra-low attachment spheroid microplates (Corning®, 4516, USA). Melanoma cells and HMEC-1 were seeded together at densities of 0.5 × 103 cells/well and 1.5 × 103 cells/well, respectively, in 40 µL of RPMI. The plate was centrifuged 500 x g for 5 min and incubated. After 24 h of incubation, either NHDF, MRC-5, or LX-2 were seeded at densities of 1.5 × 103 cells/well in a further 40 µL of RPMI, on top of the preformed spheroids, the plate was then centrifuged 500 x g for 5 min and incubated at 37 °C and 5% CO2 for 72 h.
2D and 3D DRC and IC50 determination
Generation of DRCs and determination of IC50 values of drugs in 2D tested in non-cancerous cells (NHDF, MRC-5, LX-2, and HMEC-1) were performed as follows: cells were seeded in a 96-well black plate (µClear Greiner®, Belgium) at a density of 5 × 103 cells/well in 100 µL of cell line-specific medium. Drugs were diluted in a 3-fold dilution series for 8 dilutions, with starting concentrations of Daunorubicin HCl and Pyrvinium Pamoate of 10 µM. Cell viability was determined with the CellTiter-Glo® 3D Cell Viability Assay (Promega, USA). Upon 5 days of treatment, a microplate reader Cytation 5 Cell Imaging Multi-Mode Reader (Agilent BioTek, USA) was used for luminescence measurements. The IC50 experiments were performed in technical and biological triplicates. Dose-response curves and IC50 values were generated with GraphPad 10.3.1 software (GraphPad, USA) and determined with the non-linear log (inhibitor) vs. response-variable slope (four parameters) equation. For selected melanoma cells the determination of IC50 values of drugs tested was performed in 3D as follows: cells were seeded in 384-well U-bottom ULA plates (S-Bio®, MS-9384UZ, Japan) at densities of 0.5-1 × 103 cells/well in 80 µL/well, centrifuged at 500 x g for 5 min, and incubated for 4 days at 37 °C and 5% CO2. Drugs were diluted in a 3-fold dilution series for 10 dilutions, with starting concentrations of Daunorubicin HCl of 10 µM and Pyrvinium Pamoate of 1 µM. Before drug and cell viability reagent were added, spheroids were visually inspected utilizing a bench-top microscope as a quality control step. After 5 days of treatment, cell viability was determined with the CellTiter-Glo® 3D Cell Viability Assay (Promega, USA). A microplate reader Cytation 5 Cell Imaging Multi-Mode Reader (Agilent BioTek, USA) was used for luminescence measurements. The IC50 experiments were performed in technical and biological triplicates. Dose-response curves and IC50 values were generated with GraphPad 10.3.1 software (GraphPad, USA) and determined with the non-linear log (inhibitor) vs. response-variable slope (four parameters) equation.
3D synergy assay
SKmel30 and SKmel147 cells were seeded at a density of 0.5 × 103 cells/well in 384-well ULA plates (S-Bio®, MS-9384UZ, Japan) and spheres were allowed to form for 4 days before addition of drugs. They were treated for 5 days with either single drugs or combinations of Trametinib and either Pyrvinium Pamoate or Daunorubicin HCl in a matrix format at a fixed 1:2 dilution range. Drug concentrations were pre-determined based on each inhibitor’s IC50 value. Cell viability was assessed with the CellTiter-Glo® 3D Cell Viability Assay (Promega, USA). Synergy scoring was determined using the “inhibition readout” (calculated as “100 - Cell Viability”) of the online SynergyFinder software version 3.0 (https://synergyfinder.fimm.fi) and implementing the ZIP calculation method, as published before [15]. Zero Interaction Potency (ZIP) scores < − 10 and > 10 correspond to antagonist and synergistic effects, respectively.
3D proliferation kinetic and end-point assay
Kinetic (time-lapse microscopy) cell proliferation and endpoint cell viability, under drug treatments, were evaluated as described before [11]. In brief, either mono- or -multicomponent spheroids were generated as previously described using labeled cells to allow the tracking of the different cell types. After spheroid generation, 40 µL medium were removed from each well and replaced with 40 µL medium supplemented with 2 times concentrated compounds and controls. The plate was centrifuged at 500 x g for 5 min and placed in an incubator (BioSpa8, Agilent BioTek, USA) connected to an automated live-cell imaging system (Cytation 10, Agilent BioTek, USA). Images were acquired every 12 h for 5 days using a 10x magnification objective and 590 nm LED and a Texas Red filter cube (Excitation 586/15 nm, Emission 647/57 nm) to track melanoma fluorescence signal over time. On day 5, spheroid cell viability was determined using the CellTiter-Glo® 3D Cell Viability Assay (Promega, USA). A microplate reader Cytation 5 Cell Imaging Multi-Mode Reader (Agilent BioTek, USA) was used for luminescence measurements. Kinetic and end-point cell proliferation data were analyzed and plotted with GraphPad 10.3.1 software (GraphPad, USA).
Confocal microscopy of 3D multi-component spheroids
Confocal images of 3D multi-component spheroids were acquired using the Cytation 10 (Agilent BioTek, USA) confocal microscope with spinning disk technology. The instrument is equipped with a laser combiner (spectral range 398–643 nm) and a DAPI filter cube (Excitation 390/40 nm, Emission 442/42 nm), a GFP filter cube (Excitation 472/ 30 nm, Emission 520/35 nm), and a TRITC filter cube (Excitation 556/20 nm, Emission 600/37 nm). Pictures were acquired using a 20x magnification objective.
3D apoptosis and cell death assays using confocal microscopy
Melanoma cells were seeded in 384-well black U-bottom ULA microplates (Corning®, USA) at densities of 0.5 × 103 cells/well in 80 µL/well of medium, centrifuged at 500 x g for 5 min, and incubated for 2 days at 37 °C and 5% CO2. Upon removal of 40 µL/well of medium, drugs were dispensed 2 times concentrated in 40 µL/well of medium, centrifuged at 500 x g for 5 min, and incubated for 5 days at 37 °C and 5% CO2. The positive control, Staurosporine at 1µM concentration was added 24 h previous to the end of the assay, for strong induction of apoptosis and cell death. CellEvent™ Caspase-3/7 Detection Reagent (Invitrogen, Thermo, USA) and SYTOX™ Blue Dead Cell Stain (Invitrogen, Thermo, USA) were added and incubated at 37 °C for at least 2 h. Cytation 10 was used to acquire multiple images in z-stacking using DAPI, GFP, and TRITC filter cubes and a 20x magnification objective. Brightfield pictures were also acquired at 20x magnification. Maximum intensity projected (MIP) images were generated using Gen5 (Agilent BioTek, USA). For mCherry-expressing melanoma cell lines, the mCherry signal was used to visualize the total spheroid mass, while for non-labeled melanoma cells, brightfield images were used to visualize the total spheroid mass.
3D invasion assay
Melanoma cell lines SKmel147 and M160915 were seeded in ultra-low attachment BIOFLOAT™ 96-well plates (Facellitate, Germany) in densities of 2,5 × 103 and 5 × 103, respectively. After 3 days of spheroid formation, they were embedded between two layers of Collagen type I, containing 2 mg/ml Collagen type I (MercMillipore, Germany), 1% FCS (Gibco Thermo Fisher Scientific, Waltham, USA) in RPMI (Gibco Thermo Fisher Scientific, USA). The pH of the collagen solution was adjusted to 7.4 using 1 M NaOH. 50 µl per well of Collagen I solution was pipetted into an optically clear, black-walled 96-well plate (µClear Greiner®, Belgium) and left to polymerize for 5 min at 37 °C. Next, one spheroid per well was transferred on top of the collagen layer and immediately covered with 50 µL of collagen solution and polymerized for 15 min at 37 °C. Next, 100 µl of medium containing either 0,5% DMSO (negative control) or 2 times IC50 concentration of the drug was added on top of the collagen layer. For each experimental condition, 8 spheroids were used. Pictures were taken on day 0 (immediately after embedding) and after 3 days of collagen embedding, using Cytation 10 (Agilent BioTek, USA) manual imaging mode and 4x magnification. The area of cellular invasion was analyzed using ImageJ software (Fiji). Statistical analysis was performed using GraphPad 10.3.1 software (GraphPad, USA).
Western blot analysis
Cells were seeded in 6-well Aggrewell plates (StemCell, USA) at densities of 0.5-1 × 103 cells/well in 5 mL of medium, centrifuged at 100 x g for 5 min, and incubated for 4 days. Drugs were dispensed and cells were incubated for 3 and 5 days. Cell lysis was performed on ice with cold lysis buffer (RIPA 1X containing cOmplete phosphatase inhibitor, Roche, Switzerland), protein concentration was determined using Pierce™ BCA Protein Assay Kit (Thermo, USA), and protein lysates were further analyzed by SDS-PAGE and Western Blot. The detection of enhanced chemiluminescence signals was performed as previously described [16]. Primary antibodies used in the study were: GAPDH (1:5000, polyclonal, #G9545, Rabbit, Sigma, USA), ERK (1:1000, Rabbit, L34F12, #CST4696S, CellSignaling, USA), pERK (1:1000, Rabbit, D13.14.4E, #CST4370S, CellSignaling, USA), AKT (1:1000, Mouse, 4OD4, #CST2920S, CellSignaling, USA), pAKT (Ser473) (1:1000, Rabbit, D9E, #CST4060S, CellSignaling, USA), PRAS40 (1:1000, Rabbit, #CST2610, CellSignaling, USA), pPRAS40 (1:1000, Rabbit, C77D7, #CST2997, CellSignaling, USA), S6 (1:1000, Rabbit, 5G10, #CST2217, CellSignaling, USA), pS6 (1:1000, Rabbit, 91B2, #CST4857, CellSignaling, USA), p70S6K (1:1000, Rabbit, 49D7, #CST2708, CellSignaling, USA), pp70S6K (1:1000, Rabbit, 108D2, #CST9234, CellSignaling, USA), 4EBP1 (1:1000, Rabbit, 53H11, #CST9644, CellSignaling, USA), p4EBP1 (1:1000, Rabbit, 236B4, #CST2855, CellSignaling, USA). All primary and HRP-conjugated secondary antibodies were purchased from Cell Signaling Technology (Boston, USA).
Immunofluorescence staining
Cells were seeded in multi-chamber slides (Ibidi, Germany) at a density of 20 × 103 cells/chamber in 300 µL of medium/chamber and incubated at 37 °C and 5% CO2 overnight. Drugs were dispensed and incubated for 72 h. Cells were fixed in 4% PFA for 10 min at RT and permeabilized with 0.3% Triton X solution for 10 min at RT. Afterward, cells were washed with PBS and incubated in a blocking solution (10% FCS) for 1 h. Primary antibodies were added and incubated at 4 °C overnight. β-Catenin (1:100, Rabbit, #8480S, Cell Signalling®) and γH2AX (1:200, Mouse, #80312, Cell Signalling®). Secondary antibodies conjugated with a fluorophore Alexa Fluor 488 (goat anti-rabbit, 1:500, Thermo), Alexa Fluor 647 (donkey anti-rabbit, 1:500, Thermo), Phalloidin Alexa 647 (Thermo), and DAPI (1:1000) were added and incubated for 1 h at RT. Confocal images were acquired using Cytation 10 with a 60x objective, and with DAPI, GFP and CY5 filter cubes.
Hydrogel-embedded melanoma co-culture
Melanoma-TME hydrogel encapsulation co-cultures were generated using transglutaminase cross-linkable poly (ethylene glycol) (PEG) hydrogels previously described [17]. A ready-to-use kit consisting of frozen aliquots of the 3% PEG precursor solution (8-arm 40 kDa PEG macromers bioconjugate with RGD adhesion and MMP-cleavable peptide motives) and of the activated Human Factor XIII (FXIIIa) were purchased (Ectica Technologies, Switzerland). The cell suspension was created by mixing mCherry-expressing melanoma cells at a density of 2–4 × 104/100µL with HMEC-1 expressing BFP at a density of 20 × 104/100µL, and with either NHDF, or MRC-5, or LX-2 expressing GFP at a density of 20 × 104/100µL, centrifuged at 300 x g for 3 min and supernatant was removed, and 45 µL of complete RPMI were added. Afterwards, 43 µL of PEG precursor solutions were added and gently mixed to dissolve the cellular pellet. Then, 12 µL of FXIIIa was added, and the solution was gently mixed without introducing bubbles. 5 µL of solution was dispensed in each well in a black 96-well plate (µClear Greiner®, Belgium) to create homogeneous domes and incubated at RT for 5 min until polymerization. 200 µL/well of RPMI supplemented with 10ng/mL of VEGF (Peprotech, USA) was dispensed in each well and incubated for 3 days at 37 °C and 5% CO2. 2X concentrated drugs were added in 100µL/well of fresh medium, upon removal of 100 µL/well of the old medium, and further incubated for 5 days at 37 °C and 5% CO2. The confocal microscope Cytation 10 (Agilent, BioTek, USA) was used to acquire multiple images in z-stack modality using DAPI, GFP, and TRITC filter cubes and a 20x magnification objective, selecting 4 ROIs per well. Maximum intensity projected images were analyzed using ImageJ (Fiji).
Zebrafish husbandry, determination of maximum tolerated concentrations of drugs, and xenografts
Zebrafish experiments were performed in two different institutions, the Zebrafish Facility of the University of Padova (under Italian Ministry of Health Authorization n. 1111/2024-PR (OPBA prot. D2784.185)) and the Aquatic Platform of the Luxembourg Centre for Systems Biomedicine at the University of Luxembourg (RRID: SCR_025429), in collaboration with Professor Natascia Tiso and Dr. Maria Lorena Cordero-Maldonado, respectively. Adult nacre zebrafish lines were housed in each facility according to standard protocols [18, 19]. Embryos were obtained by natural spawning and reared until the experiments at 2 dpf in E3 medium at 28° C. First, to determine the maximum tolerated concentration (MTC) of the drugs to be tested in the xenografts (Trametinib, DH and PP), we first treated non-injected naïve 2 dpf nacre larvae with serial dilutions of drugs of interest to determine the highest tolerated and non-toxic concentration until 5 dpf. Larvae viability and development were monitored daily during drug treatment. The cut-off of 20% mortality and no developmental defect was set to determine the MTC. Second, for the performance of the cell transplantations, on the day of the injections, the 2 dpf embryos were manually dechorionated and anesthetized with buffered tricaine (80 mg/l, Sigma-Aldrich). SKmel147-mCherry and MelJuso-RES-mCherry cell lines were detached using phenol red-free TryplE reagent (Gibco Thermo Fisher Scientific) and resuspended in PBS at a concentration of 2 × 105cells/ µL. The cells were injected into the yolk as a single droplet (around 100 cells per embryo) using a World Precision Instrument (Sarasota, USA) or FemtoJet 4X (Eppendorf, Germany) microinjectors. PBS with phenol red was injected as a vehicle control. After 24 h, the larvae were fluorescently assessed for successful cell implantation and subjected to drug treatment with 12 nM Trametinib, 1 µM DH, 111 nM of PP, and their combinations for 3 days at 32 °C. Larvae viability was monitored daily. After 3 days, larvae were anesthetized as described above, and photos of xenografts were taken using an M165 FC microscope with DFC7000T camera (Leica Camera, Germany) or Nikon SMZ25 fluorescent stereomicroscope (Nikon Instruments, Japan). Data was analyzed based on fluorescence intensity to measure xenograft area and number of cells using the “Measurements” tool of the Volocity 6.0 software (Perkin Elmer, Italy).
Statistical analysis
All experiments represent at least 3 biological replicates. Statistical analysis was performed using GraphPad 10.3.1 software (GraphPad, USA). The Gaussian distribution of data was assessed with Shapiro-Wilk normality test. Data following Gaussian distribution was analyzed using Ordinary one-way ANOVA with Dunett’s multiple comparison test. Data not following Gaussian distribution was analyzed using ordinary Kruskal-Wallis with Dunn’s multiple comparison test. One sample t-test was used to analyze data expressed as a percentage of the untreated control (normalized to 100%).





Comments (0)