
Remember me




At diagnosis, the patient, a right-handed 74-year-old woman, was assessed following a two-year history of behavioral changes including increased apathy, neglect of self-care, and substance use, consistent with behavioral variant FTD [1]. Motor function was intact with no signs of upper motor neuron disease or extrapyramidal/parkinsonian features (no rigidity, tremor, or bradykinesia).
Neuropsychological testing including the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) battery showed normal processing speed and attention, slight backward digit span impairment, and significant deficits in verbal learning, though recognition remained intact. Executive functions were mildly to moderately impaired, especially in inhibition and flexibility, with increased impulsivity. Language and visuo-constructive skills were within normal limits (see Table 1 for full scores).
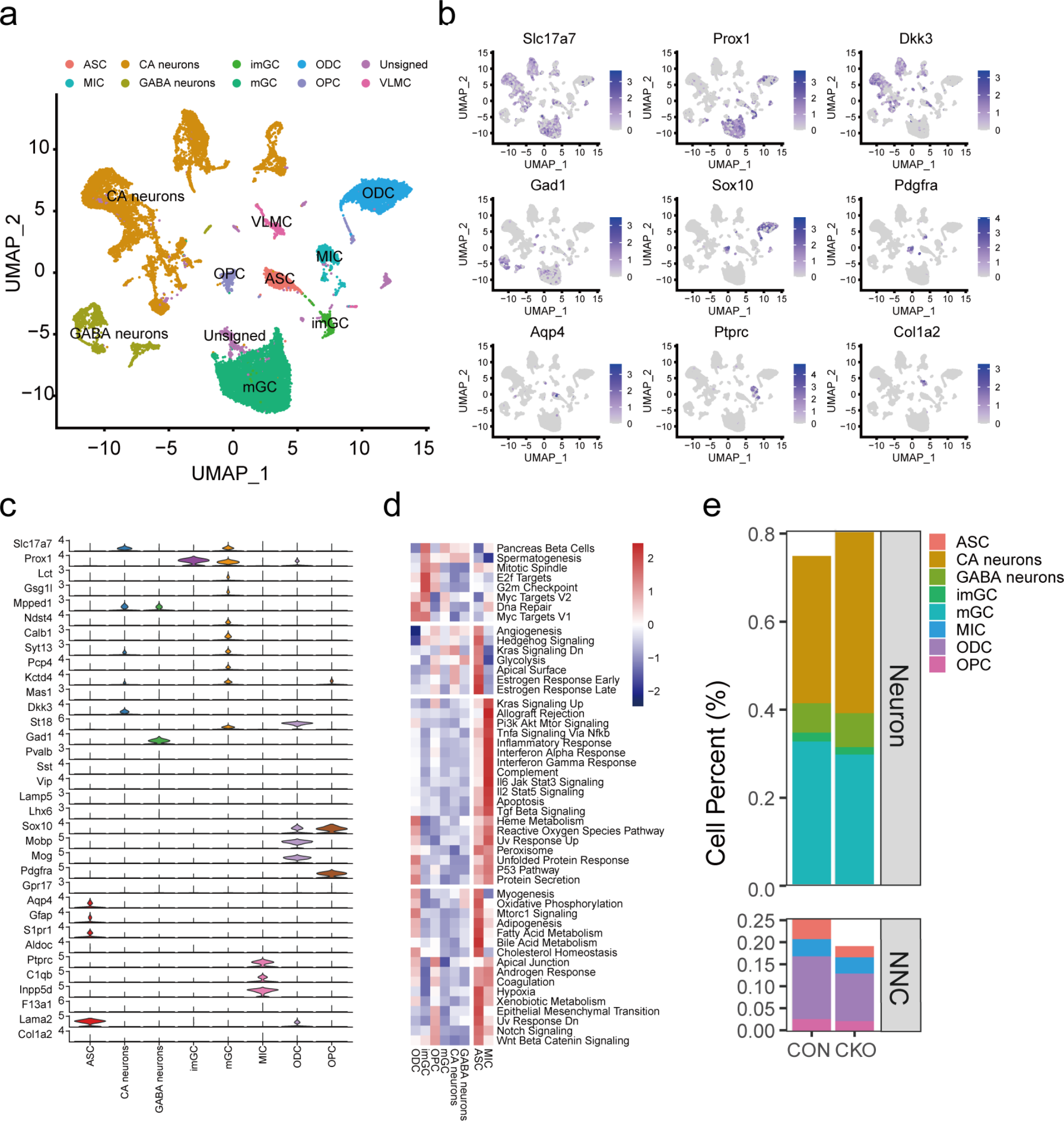
Table 1 Neuropsychological test results at baseline and one-year follow-up. Summary of raw scores and corresponding Z-scores (or percentile ranks) for key cognitive domains, demonstrating progression of memory, executive, language, and visuospatial impairments over one year. N/A indicates that the test was not administered at follow-up because the patient was too cognitively impaired to complete it. Word list recognition Z-score at follow‐up could not be calculated due to false positives. Abbreviations: WMS-R = Wechsler Memory Scale-RevisedCerebrospinal fluid (CSF) analysis was performed using the INNOTEST® ELISA platform (Fujirebio Europe N.V., Belgium) out of clinical routine CSF examination, revealing normal β-amyloid, phosphorylated tau181, and total tau levels. A positive αSyn SAA indicated the presence of misfolded aSyn aggregates suggestive of Lewy-body pathology. Using our previously validated αSyn SAA [2], CSF from 20 age- and sex-matched individuals without any clinical or biomarker evidence of neurodegenerative disease showed no seeding activity. Other CSF parameters were normal, with no evidence of infection or inflammation. The initial tau PET scan at diagnosis revealed increased [18F]PI-2620 tracer uptake in the basal ganglia, particularly on the left side with a quantitative standard uptake value ratio SUVR of 1.28, while cortical areas did not exhibit significant [18F]PI-2620 tracer uptake (Fig. 1a). Genetic analysis used a targeted dementia/FTD-ALS gene panel (APOE, APP, CHMP2B, CSF1R, DNAJC5, DNMT1, EPM2A, GRN, ITM2B, MAPT, NHLRC1, NOTCH3, OPTN, PRNP, PSEN1, PSEN2, SQSTM1, TARDBP, TBK1, TREM2, TUBA4A, TYROBP, UBQLN2, VCP), plus C9orf72 repeat expansion and APOE4 genotyping. This identified a pathogenic TBK1 mutation (c.1349_1352del (p.Ile450Lysfs*15)), consistent with autosomal dominant inheritance and resulting in TBK1 haploinsufficiency [3] - and revealed no other pathogenic or likely pathogenic variants. The patient’s three-year-younger sister was also diagnosed with a behavioral variant FTD carrying the same pathogenic TBK1 mutation, while no other family members had a history of neurodegenerative diseases.
Fig. 1
Ante-mortem detection of post-mortem confirmed pathologies as well as immunohistochemistry of presented case. (a) Ante-mortem tau PET at baseline (BL) and one year later at follow-up (FU): tau pattern imaging with standard uptake value ratios (SUVR). (b) Ante-mortem biomarkers in cerebrospinal fluid (CSF) at diagnosis indicating the absence of Alzheimer pathology (c) Ante-mortem CSF αSyn Seed Amplification Assay (SAA) at BL indicating the presence of Lewy-body (LB) pathology in the TBK1-FTD case (yellow curve) versus absence of signal in controls without clinical and biomarker evidence of neurodegenerative disease (blue curve, n = 20). Relative fluorescence unit (RFU) values are normalized to the maximum intensity of fluorescence of the respective experimental plate. Each curve represents the average of the fours replicates ± standard error of the mean. (d) TDP-43 pathology (clone 1D3) of the left frontal cortex (e) Moderate number of Lewy bodies and Lewy neurites (clone 42) in the left Nucleus basalis of Meynert (f) AT8 positive, mainly glial tau aggregates of the left Caudate Nucleus (g) AT8 positive, neuronal tau aggregates in the left Gyrus dentatus consistent with argyrophilic grain disease (AGD) pathology
One year later, the patient exhibited marked cognitive decline, dysphagia, and significant weight loss (BMI 17.0). Neurological assessment showed brisk symmetric reflexes and ideomotor apraxia, indicative of cortical dysfunction. No parkinsonian features (rigidity, tremor, bradykinesia) emerged. At one-year follow‐up, neuropsychological reassessment documented decline across all cognitive domains. Verbal memory was severely reduced: learning and delayed recall scores were markedly lower, and recognition accuracy decreased substantially. Both semantic and phonemic fluencies were significantly lower. Executive tasks requiring cognitive flexibility, inhibition, and working memory exhibited pronounced slowing, increased errors, and failure to complete certain measures due to cognitive fatigue. Visuo‐constructive copying remained largely intact, but delayed recall of visuospatial designs was significantly impaired. These findings indicate progressive, multifocal cognitive deterioration with relative preservation of basic visuoconstruction (see Table 1). A follow-up tau PET at the one-year mark showed a slight increase in basal ganglia [18F]PI-2620 uptake to an SUVR of 1.32, with cortical tracer binding remaining within normal limits (Fig. 1b).
Post-mortem examination (three years after first symptom onset) included semiautomated immunohistochemistry on a Ventana BenchMark XT platform (Ventana/Roche, Basel, Switzerland) using primary antibodies at the following dilutions: α-synuclein (mouse monoclonal clone 42, Abcam, Cambridge, UK; 1:2,000), TDP-43 (rat monoclonal clone 1D3, in-house; 1:50), phospho-tau (AT8; mouse monoclonal pSer202/pThr205, Invitrogen/Thermo Fisher, Carlsbad, CA, USA; 1:200), RD3 (3R phospho-tau, mouse monoclonal clone 8E6/C11, Millipore; 1:800) and RD4 (four-repeat (4R) phospho-tau, mouse monoclonal clone 1E1/A6, Millipore; 1:4,000). The examination revealed:
FTLD-TDP43 (Subtype A): Widespread TDP-43 protein aggregates, including neuronal cytoplasmic inclusions and short dystrophic neurites, primarily in the frontal and temporal cortices, with involvement of subcortical regions such as the caudate nucleus, putamen, and nucleus accumbens, ventromedial thalamus, amygdala, entorhinal cortex.
LBD (McKeith: limbic-stage, Braak stage 5): mild αSyn pathology according to Braak-stage 5.
Tau-pathology: neuronal and glial pathology regarding shape and distribution pattern characteristic of AGD (Saito 4), ARTAG, PART (Braak&Braak II of VI).
No detectable β-amyloid plaques (Thal-Phase 0).
Secondary findings included mild atherosclerosis and scattered microinfarcts. A summary of the ante-mortem biomarker and imaging results alongside representative post-mortem immunohistochemical findings is shown in Fig. 1.
Comments (0)