
Remember me
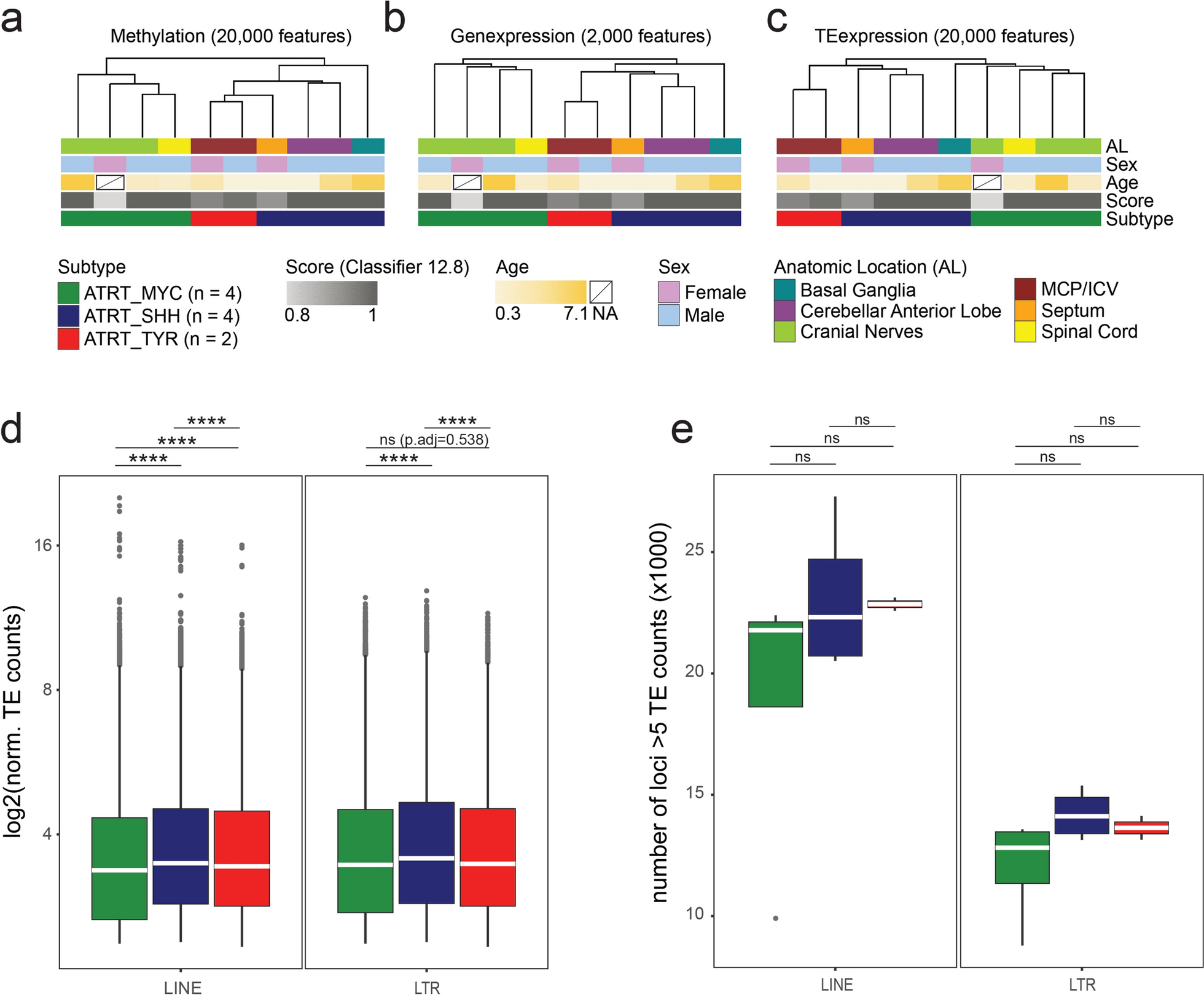



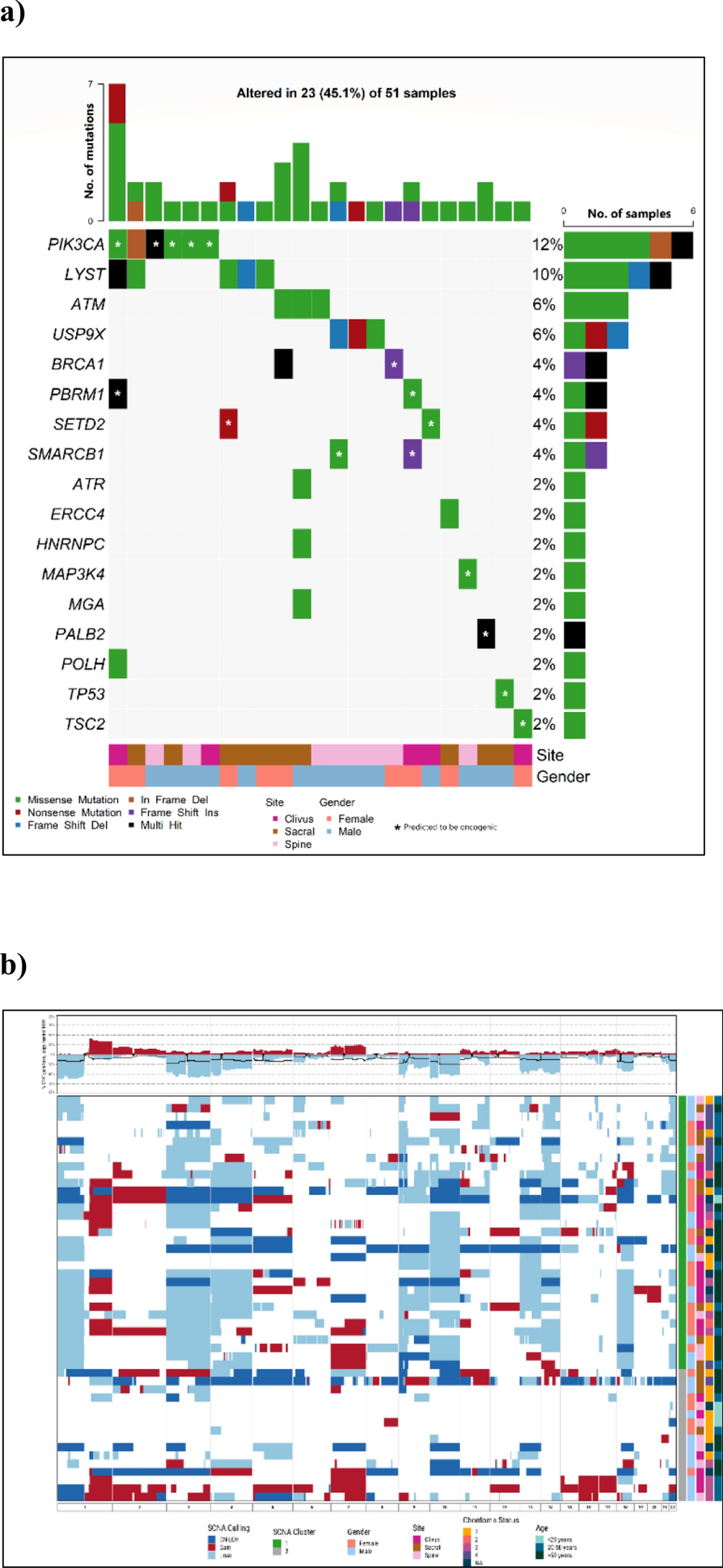

Table 1 shows the clinical characteristics for 184 chordoma patients from the US and Canada included in this study. The average age at diagnosis was 45.5 years (range 5–78); the majority were females (56.3%) and non-Hispanic Whites (95.5%). The chordoma site distribution was 49.2% clivus, 26.2% spinal, and 24.0% sacral. Consistent with previous studies [8], we found that clival patients were associated with younger age at diagnosis (Mean = 41.8 years) than patients with sacral tumors (Mean = 52.3 years, P ≤ 0.0001). Twenty-one out of 155 patients (13.5%) reported having a history of other cancer either before (N = 12) or after chordoma diagnosis (N = 7). Only breast, prostate, and skin cancer were reported by more than one patient (Table 1). Eighty-eight patients (56.8%) reported having a family history of cancer among first-degree relatives, with breast, cervical, and skin cancer the most common cancer types among female relatives, and prostate, skin, and lung cancer the most prevalent among male relatives (Table 1). Two patients were subsequently found to have family members diagnosed with chordoma. All patients were alive at the time when the initial questionnaire was completed.
Table 1 Patient characteristics in 184 chordoma patientsAmong 157 patients with treatment data available, the vast majority (97.5%) had surgery as their primary treatment after chordoma diagnosis, including 12 patients who received radiation prior to surgery (Table 1). The average time between chordoma diagnosis and the first surgery was 1.9 months, with almost all surgeries (97.3%) performed within a year. Most patients (85.3%) received additional treatment, including additional surgery (31.5%), proton radiation (42.5%), conventional radiation (24.4%), systemic (0.8%), and multi-modality (0.8%) treatment (Additional file 4). At the time of the administration of the initial questionnaire, which was on average 7.0 years after the first chordoma diagnosis, 76 patients reported not having chordoma anywhere, 31 had chordoma at the original site but the tumor was not growing, 26 had chordoma at the original site or elsewhere and the tumor was growing, and 20 had chordoma spreading to other sites (Table 1). Among 53 patients with a returned follow-up questionnaire 7–12 years after completing the initial questionnaire (on average 13.1 years since the first chordoma diagnosis), there were 12 reported deaths (Additional file 4), 9 patients (3 clivus, 4 spinal, and 2 sacral) reported a recurrence or relapse of chordoma (5 of them developed multiple recurrences/relapses), and 6 reported that the chordoma had metastasized to other organs (lung, abdomen, brain stem, and pancreas).
Genomic profilesAfter removing QC-failed samples at various steps, targeted sequencing, SNP array genotyping, and RNA profiling data were available for 51, 49, and 48 patients, respectively. Non-synonymous mutations were detected in 23 (45.1%) tumor samples, involving 17 potential chordoma driver genes (Fig. 1a). The most frequently mutated gene was PIK3CA, mutations in which were present in six (12%) patients, followed by LYST (10%), ATM (6%), and USP9X (6%). A notable observation was the seemingly mutually exclusive nature of mutations among LYST, ATM and USP9X as well as PIK3CA, ATM, and USP9X, although these are based on small numbers, within this patient cohort. Additionally, mutations within the chromatin remodeling genes PBRM1, SETD2, and SMARCB1, which have previously been recognized as frequently altered in chordoma [3, 5], were each identified in two patients. Using the CGI tool (https://www.cancergenomeinterpreter.org/home), we found that 53% of mutations shown in Fig. 1a were predicted to be driver rather than passenger mutations. Notably, almost all mutations in PIK3CA (except one), PBRM1, SETD2, SMARCB1, TP53 and MAP3K4 were predicted as driver mutations. One patient, who was a Hispanic female diagnosed with skull-base chordoma at the age of 26 and later developed metastases post-surgery and radiation therapy, exhibited both a missense mutation in PBRM1 and a frameshift mutation in SMARCB1, both predicted as driver mutations. Consistent with previous findings that TP53 mutations are rare in chordoma [3, 5], only one patient carried a TP53 mutation, which was predicted as a driver mutation based on the CGI framework (Fig. 1a). In contrast, none of the mutations observed in LYST, ATM, ATR, and USP9X were predicted as driver mutations (Fig. 1a).
Fig. 1
Genomic landscape of chordoma tumors: A Driver gene mutations; and B Somatic copy number alterations (Red: gain; Light blue: deletion; Dark blue: copy neutral LOH). Patients were clustered into two major groups. C1 (green): extensive SCNAs; C2 (gray): few or scattered SCNAs
Somatic SCNAs were identified in the majority of tumors, with the percentage of genome affected by SCNAs ranging from 1 to 70% (mean = 30% across all patients). Frequent chromosome-level or arm-level SCNAs (i.e., 90% of the p or q arm of the chromosome covered by SCNAs) included gains of chromosomes 1q, 2, and 7, and deletions of 1p, 3, 4, 9p, 9q, 10, 13q, 14q, 18, and 22 (Fig. 1b). Clustering analysis revealed two distinct groups of patients based on SCNA events. The first group (C1, N = 33) demonstrated extensive SCNAs, while the tumors in the second group (C2, N = 16) had either few SCNAs or scattered events such as copy neutral loss of heterozygosity (LOH) events and copy number gains (Fig. 1b). Amplification of the 6q27 region, which harbors the chordoma susceptibility gene TBXT, was detected in eight patients (16.3%), six of whom exhibited high copy number gains.
We utilized a NanoString panel, comprising 21 of the most differentially expressed genes identified through an RNA-Seq analysis of skull-base chordoma samples [4], to perform molecular classification within our cohort. We observed two primary groups in 48 tumors, with tumors in one of the clusters (NCC2) showing upregulation of most examined genes, as previously described in Fig. 1 of our previously published work by Bai et al. [4].
Genomic profiles in relation to clinical characteristics and outcomesAmong 63 distinct patients with any genomic data, the chordoma site distribution was 42.8% clivus, 28.6% spinal, and 28.6% sacral. The self-reported chordoma status distribution was: having no chordoma present anywhere (N = 24, 49.0%), having chordoma at the original site but the tumor was not growing (N = 5, 10.2%), having chordoma at the original site or elsewhere and the tumor was growing (N = 10, 20.4%), or having chordoma spreading to other sites (N = 10, 20.4%). Older patients (≥ 50 years) were more likely to have a higher percent of the genome affected by SCNA events (PGA, P = 0.005), particularly deletion events on chromosomes 1p, 3p, 4p, 4q, 10p, 10q, and 18q (Table 2). Interestingly, deletions of chromosome 4p were more prevalent among females than males (P = 0.004), while 7p gain was more common among males than females (P = 0.029, Table 2). Among 49 patients with both genomic and clinical data, 21 had clival, 15 had sacral, and 13 had spinal chordoma. After adjustment for age and sex, sacral tumors (OR = 5.76, P = 0.020) were more likely to harbor driver gene mutations compared to clival tumors. Deletions of chromosomes 5p (P = 0.033), 5q (P = 0.026) and 9p (P = 0.004) were more prevalent in sacral tumors compared to clival tumors, whereas chromosome 1q gain (P = 0.013) was more likely to be present in clival tumors (Table 2, Fig. 2). Among the tumors with TBXT amplification, two were clival, one was spinal, and the remaining five were sacral and from female patients. RNA expression subtype did not appear to be associated with age, sex, or chordoma site (Table 2). The individual diagnosed with a poorly differentiated chordoma had SNP array data exclusively accessible and was categorized into cluster C2 according to SCNA events observed in his tumor.
Table 2 Associations of genomic features with clinical characteristics and outcome in 49 patientsFig. 2
Distributions of genomic features by tumor location (NClivus = 21), (NSacrum = 14), (NSpine = 14)
Compared to patients free of chordoma at the time of the questionnaire, patients with persistent chordoma or who died were more likely to have deletions of chromosome 4p (OR = 13.28, 95% CI 1.11–158.60, P = 0.041), 14q (OR = 13.73, 95% CI 1.96–96.02, P = 0.008), and 18p (OR = 13.68, 95% CI 1.77–105.89, P = 0.012, Table 2, Additional file 5). On the other hand, RNA expression subtype, SCNA cluster, PGA, or driver gene mutations were not significantly associated with the presence of chordoma (Table 2). We also conducted a sensitivity analysis comparing patients with chordoma that was growing or metastatic chordoma to those with either no chordoma present or no growing chordoma. The main findings did not differ significantly (Additional file 6).
Comments (0)