
Remember me







This study is a multicenter, prospective, open-label, nonrandomized, observational phase IIb clinical trial designed to determine the safety and efficacy of a UCB-MNC product delivered into the right ventricular myocardium of patients with HLHS and its variants at the time of stage II palliation surgery (Fig. 1). This study focused primarily on short- and long-term right ventricular cardiac function. Second, cardiac injury was measured through cardiac biomarkers [troponin T and B-type natriuretic peptide (BNP)], severe adverse events (SAEs), cumulative days of hospitalization, vital signs, and parents’ perceptions of their child’s overall health and developmental ability. This study was conducted at Mayo Clinic (Rochester, MN), Children’s Hospital Colorado (Aurora, CO), Children’s Hospital of Alabama (Birmingham, AL), Cincinnati Children’s Hospital Medical Center (Cincinnati, OH), Ochsner Medical Center Jefferson (Jefferson, LA), Children’s Hospital Los Angeles (Los Angeles, CA), Children’s Hospital and Clinics of Minnesota (Minneapolis, MN), Oklahoma Children’s Hospital, University of Oklahoma (Oklahoma City, OK), and Children’s Hospital of Philadelphia (Philadelphia, PA).
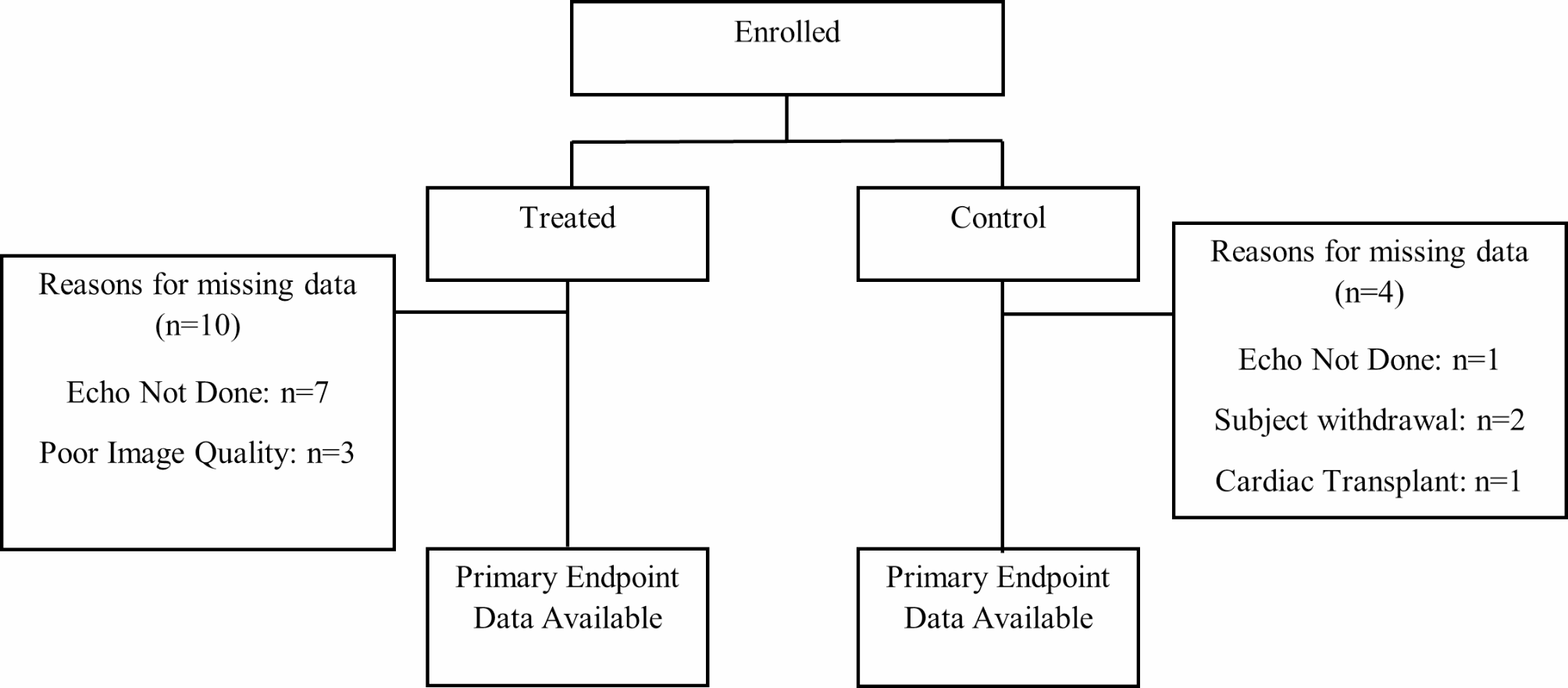
Fig. 1
Consort flow diagram showing the number of enrolled subjects, subject allocation into treatment groups, and primary endpoint data available
Eligibility and enrollmentBefore enrollment, expecting parent(s) with a prenatal diagnosis of HLHS or HLHS variants by fetal echocardiogram were enrolled in the ongoing Mayo Clinic umbilical cord blood (UCB) study for collection of UCB cells for HLHS (ClinicalTrials.gov identifier NCT01856049) under an institutional review board-approved protocol. Our study population comprised fifty subjects enrolled in the treatment group and forty-five controls. Both groups met identical study eligibility criteria, study tests, and procedures, except for the requirements related to the acceptability of the UCB-MNC product and the sensitivity of the dimethyl sulfoxide (DMSO) reaction (treatment group only). The inclusion criteria included a diagnosis of HLHS or an HLHS variant with single right ventricular-dependent CHD, a history of prior stage I palliation surgery, being scheduled for stage II palliation surgery within less than thirteen months of age after stage I surgery, and having participated in the UCB collection protocol with the autologous UCB-MNC product that was deemed acceptable for clinical use (treatment group only). The exclusion criteria included a history of DMSO, parent(s) and legal guardian(s) unwilling to have their child participate, severe chronic diseases, extensive extracardiac syndromic features, a known history of cancer, and any of the following: complications of congenital heart disease, any condition requiring urgent or unplanned interventional procedures within 15 days before stage II palliation surgery.
The study was approved by the Institutional Review Boards (IRBs) of the corresponding site institutions, which are as follows: Colorado Multiple IRB; Children’s Hospital Los Angeles IRB; Children’s Minnesota IRB; The Children’s Hospital of Philadelphia Research Institute IRB; Cincinnati Children’s Hospital IRB; Ochsner Clinic Foundation IRB; The University of Oklahoma - IRB for the Protection of Human Subjects; The University of Alabama at Birmingham - Office of the IRB for Human Written informed consent was obtained from all parents and legal guardian(s) of the subjects meeting the eligibility criteria. After providing consent, a screening phase was conducted that included a preoperative baseline evaluation: clinical evaluation, laboratory and imaging tests, and questionnaires, followed by stage II palliation surgery, which included enrollment. Subjects were actively followed up after surgery via in-clinic visits and telephone follow-ups. The protocol utilized was monitored by both the Food and Drug Administration (FDA) and the IRBs. One of our authors had full access to all the data in the study and took responsibility for its integrity and data analysis.
Manufacturing, storage, transportation, and administration of the UCB-MNC productThe autologous UCB used to manufacture the cell-based product was collected under the UCB collection study protocol at birth and shipped to ReGen Theranostics, Inc. (Rochester, MN) for manufacturing. Manufacturing this cardiac-specific product requires a unique UCB collection and cell preparation that is different from the traditional UCB collection and processing protocols that allow direct thawing and delivery at surgery. The sponsor was responsible for reviewing the release criteria on the certificate of analysis from the manufacturer for each autologous product and determining the acceptability of the product’s release to the investigational site for clinical use. During stage II palliation surgery, the frozen autologous UCB-derived product was transported to the operating room and thawed per protocol once the subject was off cardiopulmonary bypass and after anticoagulation reversal.
ManufacturingMononuclear cell enrichment was performed via Ficoll-based density gradient purification using the Sepax 2 Cell Processing System (GE Healthcare). The Sepax 2 is a closed system for cell separation, washing and concentration. Following a final centrifugation step, the UCB-MNCS were suspended in CryoStor10, a commercially available dimethyl sulfoxide-based cryopreservation solution, at concentrations ranging from 12 to 42 million cells per mL. Sterile cryovials were manually filled after at processing. The number of vials in each batch is variable and depends on cell yield. Each vial is labelled with product specific identification and a unique vial specific identifier. After processing, each product undergoes product characterization testing to meet all pre-defined release criteria. Characterization and process markers captured are hours from collection to finish, CD45, CD34, cell count, MNC total, MNC%, TNC, and viability.
Storage and transportationThe cells were stored under Good Manufacturing Practice (GMP) standards at the manufacturing facility. The products were frozen using a controlled-rate freezing process and subsequently transferred to liquid nitrogen (vapor phase) for long-term storage. For transportation, the cells were shipped in a liquid nitrogen dry shipper to the administration hospital by sponsor staff at the time of cell delivery, with no product storage at the clinical site. At the time of cell delivery, the cells are thawed by hand and immediately loaded into the delivery device with no additional manipulation. To safeguard against contamination or degradation, product delivery begins within 30 min of thawing. The procedure was canceled if a backup vial was unavailable in the surgical suite.
Product dose and administrationThe subjects in the treatment group received a target dose of 1–3 million total nucleated cells per kg of body weight of the autologous UCB-MNC product via intramyocardial injections. A 27-gauge needle attached to a 1 mL syringe was used to administer the product via direct subepicardial injections of 0.1 mL per 1 kg of body weight to reach the target dose. Injections were given slowly over at least 5 s, followed by a 20-second pause with the needle left in place to maximize cell retention within the myocardium. The injections were spaced approximately 1 cm apart across the free wall of the right ventricle. The injection sites were documented on a diagram to record the approximate location of the injection sites relative to the coronary vasculature and filed within each subject’s study file.
Cell dosing is based on subject weight, and the volume for injection to achieve the desired dose is calculated at the time of cell delivery. The target dose range was established according to experience with our preclinical studies based on variability with cell recovery in the animal models, and was used as the acceptable range for our initial phase I and the current phase II study.
The rationale for intramyocardial delivery through direct subepicardial injections was chosen based on minimizing the risk of cell delivery for these single ventricle patients. Because these patients are required to undergo open chest cardiac surgery, the epicardial surface is routinely exposed, and intramyocardial delivery at the time of surgery as an “add-on” was determined to be less invasive and less risky compared to intracoronary infusion, which would require a separate catheter-based procedure and its associated ionizing radiation. Intramyocardial injections of UCB-MNC and subsequent follow-up were conducted in preclinical studies, which confirmed the feasibility, safety, and efficacy of cell delivery.
Data acquisitionAfter consent and before stage II palliation surgery, demographic, baseline clinical, and laboratory data were collected, as were baseline quantitative measurements of cardiac function data with transthoracic echocardiography (TTE). Operative and postoperative data were also collected, including physical examination, daily vitals, and telemetry monitoring to identify arrhythmias and other adverse events; likewise, at one, three, and twelve months after surgery, physical examination, blood work-up, and TTE were performed. The Infant Toddler Quality of Life Questionnaire-97 (ITQOL-97) and the Ages and Stages Questionnaire-3 (ASQ-3) scores were also recorded at baseline and the twelve-month follow-up. The collected data were filed into applicable electronic case report forms (eCRFs) in the electronic data capture system.
Echocardiography dataTo measure our coprimary endpoints, we planned for each of the ninety-five subjects to undergo a TTE at baseline,at discharge, and at three and twelve months after surgery. Right ventricular cardiac function was measured through fractional area change (FAC) and longitudinal and circumferential strain. This study used a blinded imaging core laboratory to evaluate the echocardiography images. The imaging core lab comprises Mayo Clinic staff not affiliated with this clinical trial.
EndpointsOur coprimary efficacy endpoints assessed right ventricular cardiac function changes from baseline to three- and twelve-months following stage II palliation surgery. This assessment was conducted by measuring FAC and circumferential and longitudinal strain. The secondary endpoints assessed changes in biomarkers of cardiac injury (troponin-T, BNP) at three and six hours post surgery. Additionally, the safety endpoints included SAEs; changes in overall health, measured by vital signs (weight, heart rate, and oxygen saturation) at three and twelve months; and the cumulative incidence of hospitalization at one and three months. Finally, we measured developmental abilities at twelve months with the ITQOL-97 and ASQ-3 assessment tools following stage II palliation surgery.
Statistical analysisContinuous data are presented as medians (IQRs), and categorical data are presented as counts (%). The Kruskal‒Wallis or Wilcoxon rank sum test was used for between-group comparisons of continuous data. Categorical data were compared between groups via the chi-square test. Analysis of covariance (ANCOVA) models were used to test multiple outcomes between groups. For all ANCOVA models, we calculated the change from baseline, tested the computed change between groups, and adjusted for the baseline value of the dependent variable. Additional covariates were added in a multivariable sensitivity analysis to account for potential effect modifiers. The manuscript presents the marginal means of the ANCOVA models as LS means. Spearman correlation was used to assess the relationship between characterization and process markers and the primary outcomes. The primary and secondary endpoints were defined a priori, with additional comparisons added to better describe the sample, baseline status, and group differences in this work. An alpha of 0.05 was prespecified, and coprimary endpoints were adjusted post hoc to preserve alpha using the Bonferroni adjustment. No other adjustments were made for multiple testing outside the coprimary endpoints, and these results should be considered exploratory and not inferential. All analyses were performed via SAS Software (version 9.4), SAS Institute, Inc.
Comments (0)