
Remember me
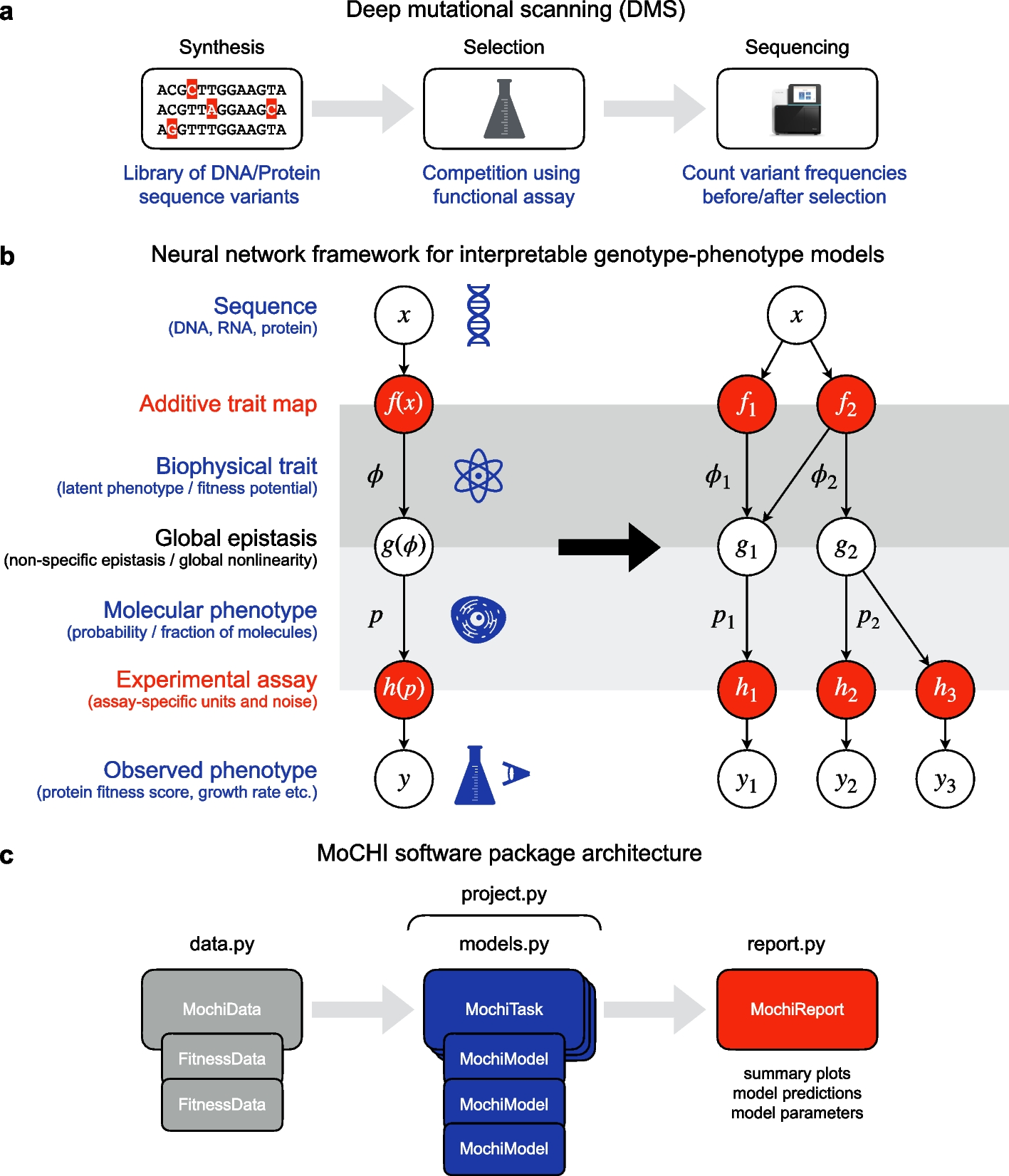
Input materials for target enrichment library were provided by the German Cancer Research Center (DKFZ), Heidelberg, Germany. Four main GBM subgroups, as defined based on sequence and methylation profiling [62], were included. Equivalent subgroup representation was given (Additional file 1: Table S5).
The Cancer Genome Atlas (TCGA)Gene expression (RNAseqV2 normalized RSEM) and DNA methylation data (HumanMethylation450) were downloaded in May 2019 using TCGAbiolinks [63,64,65] for the following cancer types: BRCA (778 genomes), CESC, (304), COAD (306), ESCA (161), GBM (50), KICH (65), KIRC (320), KIRP (273), LIHC (371), LUAD (463), PAAD (177), SKCM (103), THYM (119).
NIH Roadmap Epigenomic Project [66]H3K4me1 broad peaks of corresponded TCGA tumor types and DNaseI cell-specific narrow peaks of normal brain (E081 and E082) were obtained.
Encyclopedia of DNA Elements (ENCODE) [67]DNaseI hypersensitivity peak clusters (wgEncodeRegDnaseClusteredV3.bed.gz) and transcription factor ChIP-seq clusters (wgEncodeRegTfbsClusteredWithCellsV3.bed.gz) and DNase brain tumor data (Gliobla and SK-N-SH) were obtained. The ENCODE transcription factor binding (TFB) scores presented in Fig. 2 represent the peaks of transcription factor occupancy from uniform processing of ENCODE ChIP-seq data by the ENCODE Analysis Working Group. Scores were assigned to peaks by multiplying the input signal values by a normalization factor calculated as the ratio of the maximum score value (1000) to the signal value at one standard deviation from the mean, with values exceeding 1000 capped at 1000. Peaks for 161 transcription factors in 91 cell types are combined here into clusters to produce a summary display showing occupancy regions for each factor and motif sites within the regions when identified. One-letter code for the different cell lines is given in https://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodeDCC/wgEncodeRegTfbsClustered/wgEncodeRegTfbsClusteredV3.bed.gz.
Additional public dataHiC Data for TADs were downloaded from https://wangftp.wustl.edu/hubs/johnston_gallo/ [68].
Cell linesHuman GBM T98G cells were purchased from the ATCC collection (ATCC® CRL-1690™) and cultured in minimum essential medium-Eagle (Biological Industries), supplemented with 10% heat-inactivated FBS #04-127-1A (Biological Industries), 1% penicillin/streptomycin P/S # 03-031-1B (Biological Industries), 1% L-glutamine #03-020-1C (Biological Industries;), 1% non-essential amino acids, #01-340-1B (Biological Industries), and 1% sodium pyruvate #03-042-1B (Biological Industries), at 37 °C and 5% CO2.
GenesGenes analyzed in the study included the pan-cancer driver genes listed by Vogelstein et al. [36] including the GBM driver genes listed by Kandoth et al. [35], but excluding the HIST1, H3B, and CRLF2 genes due to missing expression data, and the AMER1 gene for which probe design failed. Cancer type-specific genes (n = 23) were selected from a published list of 840 genes [69]. Non-driver candidate GBM genes (n = 14) were suggested by BR. Non-driver variable genes (n = 22) were defined as those showing top expression variation among the 70 analyzed GBM samples for which we found at least two correlative sites in the TCGA-GBM dataset. We used the genomic coordinates for gene features from the hg19 refGene table of the UCSC Genome Browser [70]
Target enrichment assaysVariable regulatory regions were defined as the regions carrying H3K4me1 marks in all tumors, and also H3K27ac in at least 25% of the tumors, but not in at least other 25% of the tumors. RNA probes were designed to target all methylation sites within these regions, utilizing the SureDesign tool (https://earray.chem.agilent.com/suredesign/). Probe duplication was applied in cases (n = 8652) of > 5 CpG sites within the 120 bp span of the probes. Repetitive regions were identified by BLAT [71] and excluded from the design. Custom-designed biotinylated RNA probes were ordered from Agilent Technologies (https://www.agilent.com).
Genomic tumor DNAs were arbitrarily sheared using a sonication device into collections of DNA fragments of various sizes. These DNA segments were then allowed to attach the probes which fully or partially overlapped their span. The resulting collection of captured DNA segments (median size = 224 bp) was integrated into gene-reporting vectors or underwent sequencing.
Enrichment libraries of GBM-targeted regulatory DNA segments were constructed using the SureSelect protocol #G9611A (Agilent) for Illumina multiplexed sequencing, which used 200 nanograms genomic DNA per reaction, or the SureSelect Methyl-Seq protocol #G9651A using 1 μg genomic DNA per reaction. Quality and size distribution of the captured genomic segments were verified by TapStation nucleic acids system (Agilent) assessments of regular or bisulfite-converted libraries. Target enrichment efficiency and coverage was evaluated via sequencing.
Massively paralleled reporter assayMassively parallel functional assays were performed as described by Arnold et al. [72], with the following modifications.
Reporter backboneThe pGL3-promoter vector (Promega, GenBank accession number U47298) was modified as described in Additional file 2: Fig. S15.
Genomic inputsPlasmid libraries were constructed using a target-enriched library as input material: 1 μL of adaptor-ligated DNA fragments from the AK100 target enrichment library was amplified in eight independent PCR reactions, using KAPA Hifi Hot Start Ready Mix #KK2601 (KAPA Biosystems). Reaction conditions included 45 s (s) at 95 °C, 10 cycles of 15 s at 98 °C, 30 s at 65 °C, 30 s at 72 °C, and 2 min final extension at 72 °C, applying the forward Illumina universal primer: 5′-TAGAGCATGCACCGGTAATGATACGGCGACCACCGAGATCT-3′ and reverse Indexed Illumina primer: 5′-GGCCGAATTCGTCGACCAAGCAGAAGACGGCATACGAGAT-3′, containing Illumina adapter sequences. A specific 15-nt extension was added to each adapter as homology arms for directional cloning. PCR reactions were pooled and purified on NucleoSpin Gel and PCR Clean-up #740609 columns (Macherey–Nagel). The screening vector was linearized with AgeI-HF and SalI-HF restriction enzymes (NEB) and purified through electrophoresis and gel extraction. Purified PCR products were cloned into the linearized vector by recombination with the adapter-ligated homology arms in 12 reactions of 10 μL each, applying the In-Fusion HD #639649 kit (Clontech). The reactions were then pooled and purified with 1x Agencourt AMPureXP DNA beads #A63881 (Beckman Coulter) and eluted in 24 μL nuclease-free water.
Library propagationAliquots (n = 12, 20 μL each) of MegaX DH10B T1 Electrocomp Bacteria #C640003 (Invitrogen) were transformed with 2 μL of the plasmid DNA library, according to the manufacturer’s protocol, except for the electroporation step, which was performed using the Nucleofactor 2b platform (Lonza) Bacteria program 2. Every three transformation reactions were pooled (total of 4 reactions) for a 1-h recovery at 37 °C, in SOC medium, while shaking at 225 rpm, after which each reaction was transferred to 500 ml LB AMP (Luria Broth Ampicillin) for overnight 37 °C incubation, while shaking at 225 rpm. Propagated plasmid libraries were extracted using the NucleoBond Xtra Maxi Plus Kit (#740416) (Macherey–Nagel). To verify unbiased amplification of the targeted genomic segments, the size distribution and coverage of the library were analyzed before and after the propagation step (Additional file 2: Fig. S16).
In vitro methylation assayComplete de-methylation stages were achieved by propagation of the libraries in bacteria following PCR amplification stages. In vitro methylation of the de-methylated plasmid DNA was performed using the New England Biolabs CpG Methyltransferase M.SssI #M0226M according to the manufacturer’s instructions. Efficient methylation level was confirmed by using a DNA protection assay against FastDigest HpaII #FD0514 (Thermo Scientific) digestion (Additional file 2: Fig. S17).
Transfection to GBM cellsTwenty micrograms of DNA were transfected into 2 × 106 T98G and U87 cells at 70–80% confluence, using the Lipofectamine 3000 transfection kit #L3000-015 (Invitrogen), according to the manufacturer’s protocol. In each experiment, 5 × 107 cells were transfected and incubated at 37 °C, for 24 h.
Isolation of plasmid DNA and RNA from GBM cellsPlasmid DNA was extracted from 2.5 × 107 cells, 24 h post-transfection. Cells were rinsed twice with PBS pH 7.4, using the NucleoSpin Plasmid EasyPure kit #740727250 (Macherey–Nagel), according to the manufacturer’s protocol. Total RNA was extracted from 2.5 × 107 cells 24 h post-transfection using GENEZOL reagent # GZR200 (Geneaid), according to the manufacturer’s protocol. The polyA + RNA fraction was isolated using Dynabeads Oligo-(dT)25 #61002 (Thermo scientific), scaling up the manufacturer’s protocol 5-fold per tube, and treated with 10 U turboDNase #AM2238 (Invitrogen) at 20 ng/μL 37 °C, for 1 h. Two reactions of 50 μL each were pooled and subjected to RNeasy MinElute clean up kit #74204 reaction (Qiagen) to inactivate turbo DNase and concentrate the polyA + RNA.
Reverse transcriptionFirst-strand cDNA synthesis was performed with 1–1.5 μg polyA + RNA in a total of four reactions, 20 μL each, using the Verso cDNA Synthesis Kit #AB1453B (Thermo Scientific) according to the manufacturer’s protocol, with a reporter-RNA specific primer (5′-CAAACTCATCAATGTATCTTATCATG-3′). cDNA (50 ng) was first amplified by PCR, at 98 °C for 3 min, followed by 15 cycles at 95 °C for 20 s each, 65 °C for 15 s, and 72 °C for 30 s. Final extension was performed at 72 °C for 2 min, using the Hifi Hot Start Ready Mix (KAPA), with reporter-specific primers. Forward primer: 5′-GGGCCAGCTGTTGGGGTG*T*C*C*A*C-3′ which spans the splice junction of the synthetic intron, and reverse primer: 5′-CTTATCATGTCTGCTCGA*A*G*C-3′, where “*” indicates phosphorothioate bonds. In total, 16–20 reactions were performed. The amplified products were purified with 0.8 × Agencourt AMPureXP DNA beads and eluted in 20 μL nuclease-free water. The resultant purified products served as a template for a second PCR performed under the following conditions: 95 °C for 3 min, 12 cycles of 98 °C for 15 s, 65 °C for 30 s, 72 °C for 30 s. Final extension was performed at 72 °C for 2 min, with forward Illumina universal primer: 5′-TAGAGCATGCACCGGTAATGATACGGCGACCACCGAGATCT-3′ and reverse indexed Illumina primer: 5′-GGCCGAATTCGTCGACCAAGCAGAAGACGGCATACGAGAT-3′. PCR products were purified with 0.8 x Agencourt AMPureXP DNA beads, eluted in 10 μL nuclease-free water, and pooled.
Transcriptional activity analysisQuality and size distribution of extracted plasmid DNAs and RNAs were verified using TapeStation (Additional file 2: Fig. S18). DNA and cDNA samples were sequenced using the HiSeq2500 device (Illumina), as per the 125-bp paired-end protocol. Alignment with the hg19 reference genome was performed on the first 40 bp from both sides of the DNA segments, using Bowtie2 [73]. Reads with mapping quality value above 40 aligned with the probe targets were considered for further analyses. Each of the captured genomic segments was given a unique ID according to genomic location and indicated the total number of DNA and RNA reads. Only on-target segments with at least one RNA read (n = 623,223 pre-methylation; 304,998 post-methylation) were included. > 99% of the targeted regions were presented following the propagation in bacteria and re-extraction from T98 cells (Additional file 1: Table S12). Technical and biological replications performed using Illumina MiSeq sequencing.
Transcriptional activity score (TAS) was calculated as follows:
$$\mathrm= }_((}_/}_)/(}_}/}_})),$$
where j is a genomic element and RNAtotal or DNAtotal is the sum of all segment reads.
For the analyses of isolated regulatory elements, TAS was determined in 500 bp, 50% overlapping windows, across the genome, based on the DNA and RNA reads of segments overlapping with the given window. TAS significance was tested by chi-square against total RNA to DNA. Multiple comparisons were corrected. Functional regulatory elements were defined as elements with FDR q value < 0.05 and minimum 100 RNA reads, where positive TASs were defined as enhancers and negative as silencers. The methylation effect was analyzed by calculating TAS difference between treatments, where regulatory elements with a difference of ≥ 1.5-fold activity were counted.
Inferring cis-regulatory circuitsMethylation sequencingMethyl-seq-captured libraries were sequenced using a Hiseq2500 device (Illumina), by applying paired-end 125 bp reads. Sequence alignment and DNA methylation calling were performed using the Bismark V0.15.0 software [74] against the hg19 reference genome. The sequencing yielded 52–149 million reads per sample, at an average mapping efficiency of 78.1%, average bisulfite efficiency of 97.6%, and 99.4% on target average. Overall, a mean coverage of 916 reads per site was obtained, and 86% of the targeted sites were covered by at least 100 reads. Sites that appeared in less than eight of the tumors were excluded from the analyses.
Circuit annotationCorrelation between the expression level of each targeted gene and the DNA methylation level of targeted CpG sites in a 2-Mbp region flanking its transcription start site (TSS) was assessed by applying pairwise Spearman’s rank correlation coefficient with Benjamini–Hochberg correction for multiple-hypothesis testing at FDR < 5%. Circuits with R2 > 0.3 were included. Sites that correlated (R2 > 0.1) with expression of the PTPRC (CD45) pan-blood cells marker were considered a possible result of blood contamination and were eliminated from later analyses, as described [75]. Potential secondary effects were considered in two cases: (1) the correlated site was included within the prescribed portion (the gene body, excluding the first 5 Kbp) of another gene, and (2) the correlated site was located within the promoter (from TSS-1500 bp to TSS + 2500 bp) of another gene. For these cases, a correlation between the expression level of the genes was tested, and circuits with R2 > 0.1 that fit one of the scenarios described in Additional file 2: Fig. S7 were excluded. For model developing, we excluded circuits which mismatched the report assay: circuits with methylation sensitive TAS (which were calculated for the DNA segments overlapping the given site and were changed by × 1.5 fold by methylation) which mismatched the canonical mode (i.e., groups I and II in Fig. 2g).
Methylation-based prediction of gene expressionFor each gene, we performed two methods: (1) multiple linear regression and (2) lasso regression. (1) In multiple linear regression, we should reduce the number of variables since we have only 24 samples. Thus, we tested all the possible combinations of one to four associated sites. For each combination with full data in at least 12 tumors, we generated a predictive model of expression level based on multiple linear regression of the sites’ methylation levels. A significant model (q value < 0.05) was evaluated by ANOVA for linear model fit and corrected for the number of possible models per gene by FDR. A gene was considered to have a synergic model if the predictive value of the model was better than each of the involved sites alone.
Validation of methylation-based predictions was performed using the leave-one-out cross validation approach for assessing the generalization to an independent dataset. One round of cross-validation involves 23 datasets (called training set) in which performing all the analysis and one sample for validating the analysis (called testing set). The cross-validation was performed 24 times. For each training dataset, cis-regulatory circuits were generated (as described in the “Circuit annotation” section), and possible predictive models were developed for the targeted genes. Prediction quality of each gene was then tested in the 24 rounds, by comparing predicted versus observed expression level. Difference up to 2-fold was considered as a success. The ability to accurately predict the expression level of a gene was considered verified if it has good prediction quality in at least 20 of the 24 rounds.
The ability to predict gene expression alterations was performed by analyzing the genes with irregular expression levels in certain tumors. Out of 4248 analysis sets (177 genes in 24 left-out tumors), 2652 were for genes with prediction models. Of them, 868 were for genes with irregular expression (expression level in the left-out tumor was > 1SD of the gene expression levels across the tumors). Predictions in which the prediction errors were within twofold of the observed expression-level were considered accurate.
Analysis of coding sequence variationsVCF files describing single nucleotide variations (SNV) were provided by the DKFZ. Synonymous SNV, SNVs overlapping with published SNPs (COMMON), or SNVs with a less than 25-read coverage or bcftools-QUAL score > 20 were excluded. Copy number variations (CNV) were analyzed by whole-genome sequencing (WGS) data provided by the DKFZ. Association between gene expression and copy number was evaluated by Pearson or Spearman’s correlations. p-values were adjusted for multiple-hypothesis testing using the Benjamini–Hochberg method, with FDR < 5%.
Analysis of regulatory sequence variationsPre-alignment processingGBM tumors (n = 8) were sequenced using the paired-end 250- or 300-bp read protocol in Illumina MiSeq V2 or V3 devices. FASTQ files were filtered, and sequence edges of Phred score quality > 20 were trimmed up to 13 bp of Illumina adapter applying Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Reads that were shortened to 20 bp or less were discarded, along with their paired read. Exclusion of both reads was implemented after verifying that retention of unpaired reads did not significantly increase high-quality alignment coverage. Quality control of the original and filtered FASTQ files was performed with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc), deployed to verify the reduction in adapter content and the increase in base quality following the filtering stage. Duplicates were removed at the pre-alignment stage with FastUniq [76]. Duplicate pair-ends were removed by comparing sequences rather than post-aligned coordinates, allowing preservation of variant information.
Sequence alignmentSequences were aligned to GRCh37/hg19 assembly of the human genome applying paired-reads Bowtie 2 [73]. Discordant pairs or constructed fragments larger than 1000 bp were discarded, thereby improving mapping quality by allowing both reads to support mapping decisions. Default values (Bowtie 2 sensitive mode) were applied to end-to-end algorithm parameters, seed parameters, and bonus and penalty figures. Outputted SAM and BAM alignment files were examined using the Picard CollectInsertSizeMetrics utility to verify correctness of final insert-size distribution (http://broadinstitute.github.io/picard; version 1.119).
Variation callingA BCF pileup file was generated from each BAM file using the samtools [77] mpileup function, set to consider bases of minimal Phred quality of 30 and minimal mapping quality of 30. Variant calling performed using bcftools was initially set to output SNPs only to create SNP VCF files, according to the recommended setting for cancer [78]. The VCF files were filtered by applying depth of coverage (DP) above 40 and statistical quality (QUAL) above 10. DP filtering in this context refers to DP/INFO in the VCF file, which is a raw count of bases.
Variant post-processingPost-processing of VCF SNPs included additional filtering, variant frequency calculation, mapping variants to probes, and mapping variants to public databases, performed with a custom-written Python script. Additional depth coverage filtering of 20 was applied on the high-quality bases, which were selected by bcftools as appropriate for allelic counts. Frequency calculations were based on high-quality allelic depth (ratio of each allelic depth to sum of all allelic depths). SNPs were mapped to the following dbSNP [79] and ClinVar [80] databases: dbSNP/common version 20170710, dbSNP/All version 20170710, and clinvar_20170905.vcf. A match was determined when the position, reference, and variant were all in agreement. In our analysis, we refer to de novo variations (not in COMMON and not in ALL) which were detected in at least one sample (of eight). For each targeted gene, we counted the number of de-novo variations that were at a distance of ± 500 bp from its correlated sites.
Regulatory CNVsNon-coding CNVs were detected from WGS of 5 Kbp sliding blocks in 2-Mbp region flanking gene TSSs, with a 50% overlap. Correlation of the total copy number (TCN) of each block with the gene expression level was assessed (at least six samples with available TCN data, Pearson and Spearman correlation). Correlation p values were adjusted for multiple-hypothesis testing using the Benjamini–Hochberg method.
Genome editingDesign and cloning of sgRNAGuides to perturb SMO regulatory units were designed using the ChopChop, E-CRISP, and CRISPOR software; 20-bp sgRNA sequences followed by the PAM “NGG” for each unit were identified and synthesized. For the SMO regulatory unit at chr7:128,507,000–128,513,000 designated unit “A,” 4 guides were cloned into a backbone vector bearing puromycin resistance (addgene, 51,133), using the Golden Gate assembly kit (NEB® Golden Gate Assembly Kit #E1601). Each guide sequence was cloned with its own U6 promoter and was followed by a sgRNA scaffold. For the regulatory unit at chr7:129,384,500–129,389,500, designated unit “D,” two guides were cloned into the same backbone plasmid using the same method (Additional file 2: Fig. S7).
Transfection/CRISPR–Cas9-mediated deletionAfter validating the sgRNA sequences by Sanger sequencing, T98G or T98GdeltaSMO-D cells were co-transfected with a Cas9-bearing plasmid (addgene, 48138) and either the plasmid bearing the guides targeting SMO A, the plasmid with bearing the guides targeting SMO D, or the same plasmid harboring a non-targeting gRNA sequence (scramble) as a negative control. The molar ratio between the transfected guide plasmid and the Cas9 plasmid was 1:3, in favor of the plasmid not carrying the antibiotic resistance. 1.5–3 × 105 cells/ml, > 90% viable, were plated 1 day prior to transfection in a 6-well dish. On the transfection day, each well received 3 μL Lipofectamine® 3000 Reagent, 5 μg total plasmid DNA, and 10 μL of Lipofectamine® 3000 Reagent (2:1 ratio). Puromycin (3 μg/μL) was added to the cells 1 day after transfection. After 72 h, the antibiotic was washed, and the cells were left to expand. The cells were harvested 8–21 days post-transfection, and genomic DNA and RNA were immediately collected (Qiagen; DNeasy #69504 and RNeasy #74106, respectively).
Genotyping of mutant populationsGenomic DNA was subjected to genotyping PCR (primers listed in table). Deletion or partial deletion was confirmed by gel electrophoresis or TapeStation, by Sanger sequencing, and by Illumina MiSeq sequencing (150 bp paired-end). Sanger sequencing analyzed using BLAST® and sequence logo was generated using by ggseqlogo R package [81]. RNA extracted from populations of cells bearing such mutations was then checked for an effect on SMO transcription level, using qPCR (QuantStudio 3 cycler, Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA).
Single-cell dilution to obtain CRISPR-targeted cell clonesPuromycin-selected cells were isolated by trypsinization, counted and diluted to a concentration of 20 cells/100 μL. Diluted cells (200 μL) were then serially diluted, to ensure single-cell occupancy of rows 6–8 (eight dilution series). By calibrating the number of cells in the first row, we ensured that single cells could be isolated from the sixth to eighth rows onwards. Cells were incubated until the low-density wells were confluent enough to be transferred to 24-, 12-, and finally to 6-well plates. Selected clones were tested for a stable DNA profile and for SMO transcription level by genotyping PCR (primers listed in table), followed by gel electrophoresis or TapeStation and qPCR analysis, respectively.
RT-qPCREach isolated mRNA (500 ng) was transcribed to cDNA using the Verso cDNA Synthesis Kit (#AB-1453/A, Thermo Fisher Scientific, Waltham, MA, USA) according to provided instructions, using the oligo dT primer. qPCR was performed using the Fast SYBR™ Green Master Mix (#AB-4385612, Thermo Fisher Scientific, Waltham, MA, USA) and qPCR primers for SMO and reference genes HPRT and TBP (see table), on a QuantStudio 3 cycler (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA). The reaction was conducted in triplicates, and 20 ng of template was placed in each well. For each primer set, a no-template control (NTC) was also run, to check for possible contamination. QuantStudio Design & Analysis Software v1.4.3 (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) was used for analysis. All presented data were based on three or more biological replications of the genome editing experiments, each with three technical repeats of the DNA and RNA.
Guide list
A1
ACCCTGCGCGCCGAGGTATC
A2
GCGACCTGGGAGCCGCCGCC
A3
ACCGCCGGTGCCGACCTTTG
A4
GCGTGGTAGTCCTTCTCCGG
D1
GTCCTGCTCTATCTTGTCGT
D2
CACATGTAGGTCTTTCTGAC
N1
CCGGCTCTGGGACTTACACCAATG
N2
CCGGACGGTGGATCTTCTTTAGTT
N3
CCGGTCCACCTTTTTGTTTCCTCT
N4
CCGGAAGATGGATGTCCCAGCACC
Primer list
Genotyping SMO A (F)
1066F
GCAGTGCGCTCACTTCAAA
Genotyping SMO A (R)
1066R
CTCCTGGGGCGAGATCAAAG
Genotyping SMO D (F)
1069F
CATGGTCCCGGTTCCCATTTGG
Genotyping SMO D (R)
955R
GCCCTCCACAGACCAAACAGC
Genotyping SMO NULL (F)
1120F
GCTCAGTCTCAGTGTGGGAG
Genotyping SMO NULL (R)
1120R
GGCGTTTCCACAAGAGATGAGC
qPCR SMO F
950F
TGCTCATCGTGGGAGGCTACTT
qPCR SMO R
950R
ATCTTGCTGGCAGCCTTCTCAC
qPCR HPRT F
442F
TGACACTGGCAAAACAATGCA
qPCR HPRT R
442R
GGTCCTTTTCACCAGCAAGCT
qPCR TBP F
850F
TGCACAGGAGCCAAGAGTGAA
qPCR TBP R
850R
CACATCACAGCTCCCCACCA
Statistics and data visualizationAll analyses were performed using both public and custom scripts written in R (http://www.R-project.org) and MATLAB (The Mathworks, Inc.). Plots were generated using plotting functionalities in base R and using ggplot2 package (https://ggplot2.tidyverse.org) and corrplot package (https://github.com/taiyun/corrplot). Sequence logos were generated using the ggseqlogo package [81]. Heatmaps were produced using the ComplexHeatmap package [82]. Lasso regression was performed using the default parameters of gmlnet package [83].
Comments (0)