
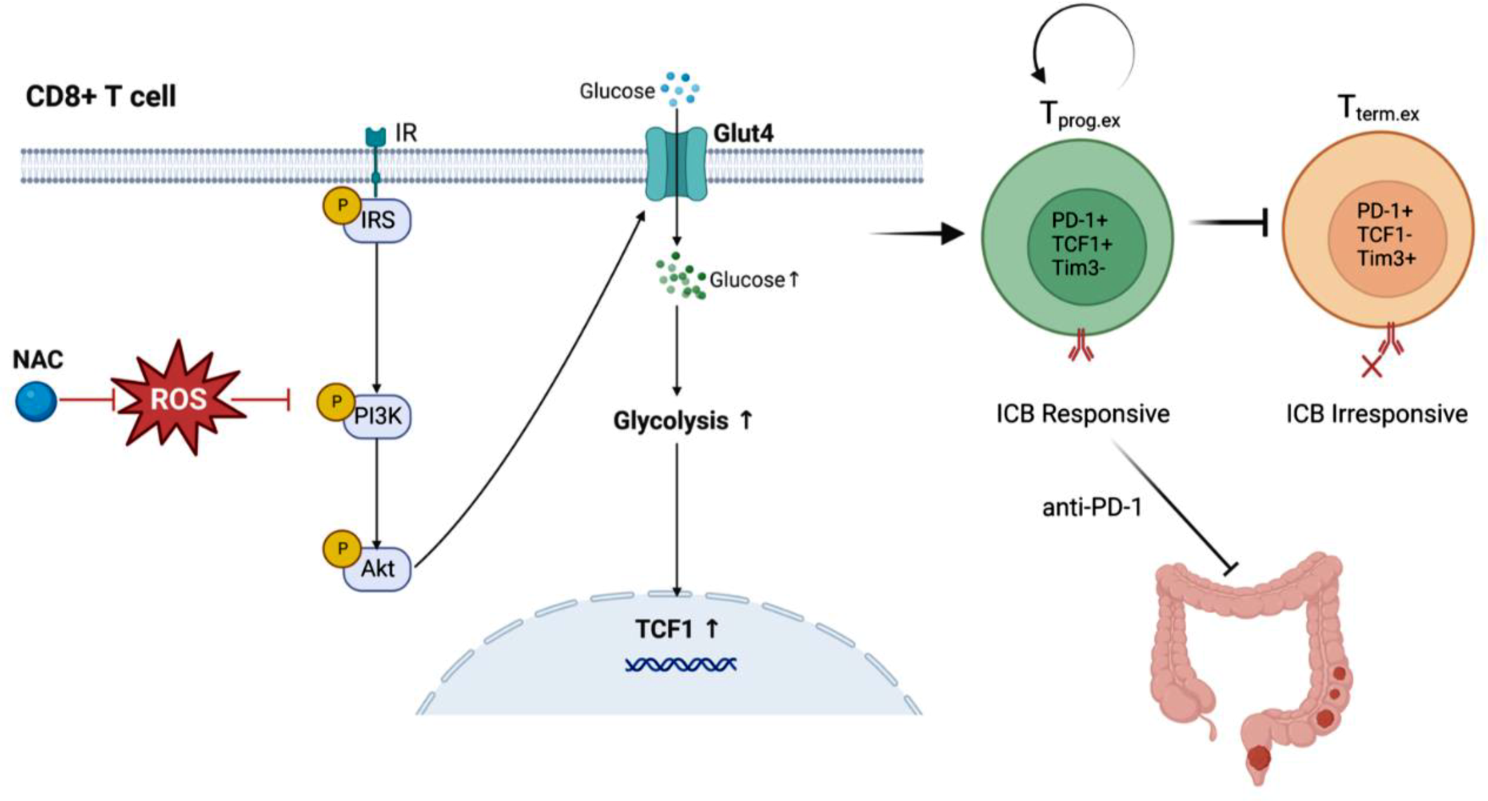
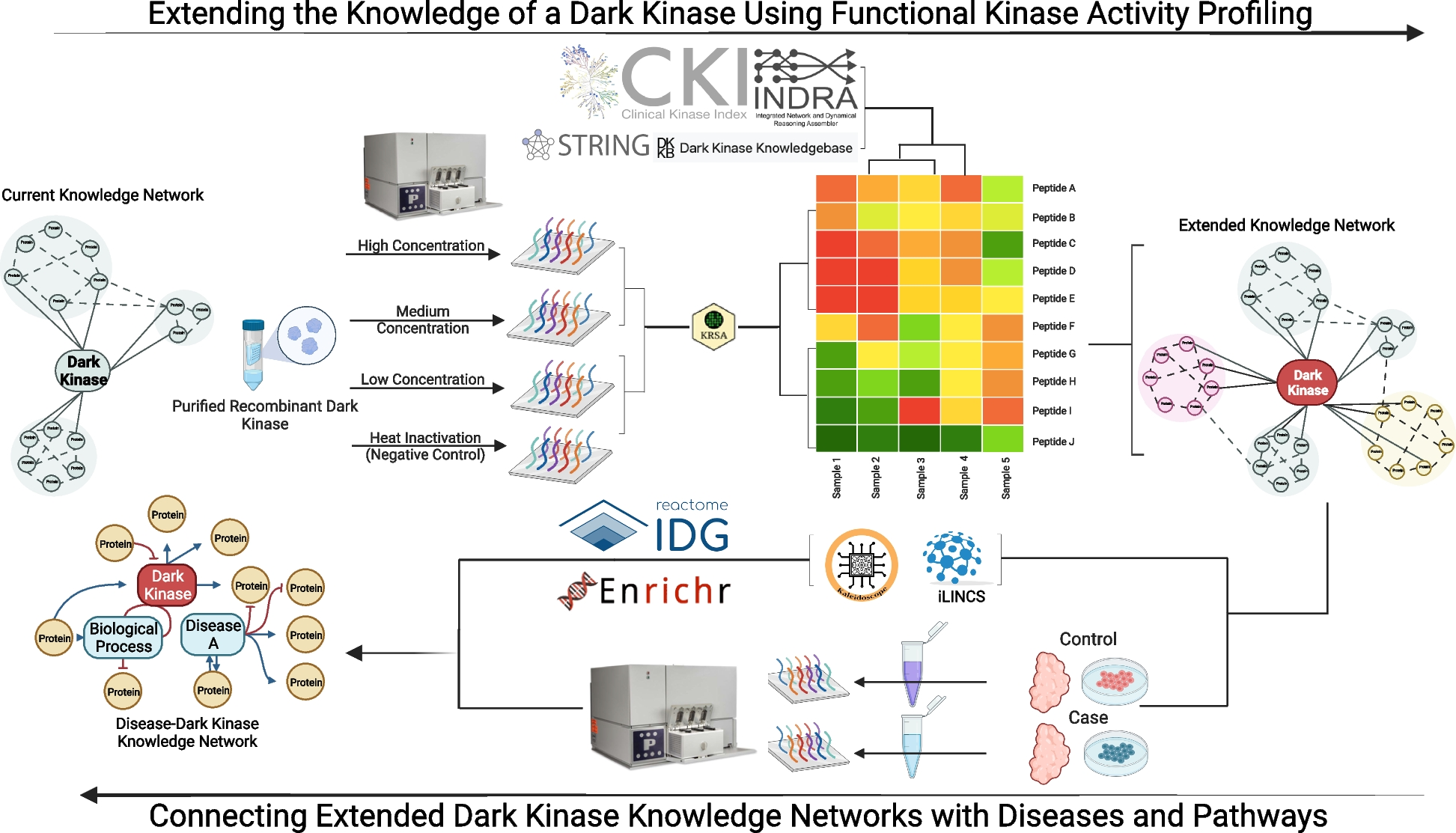
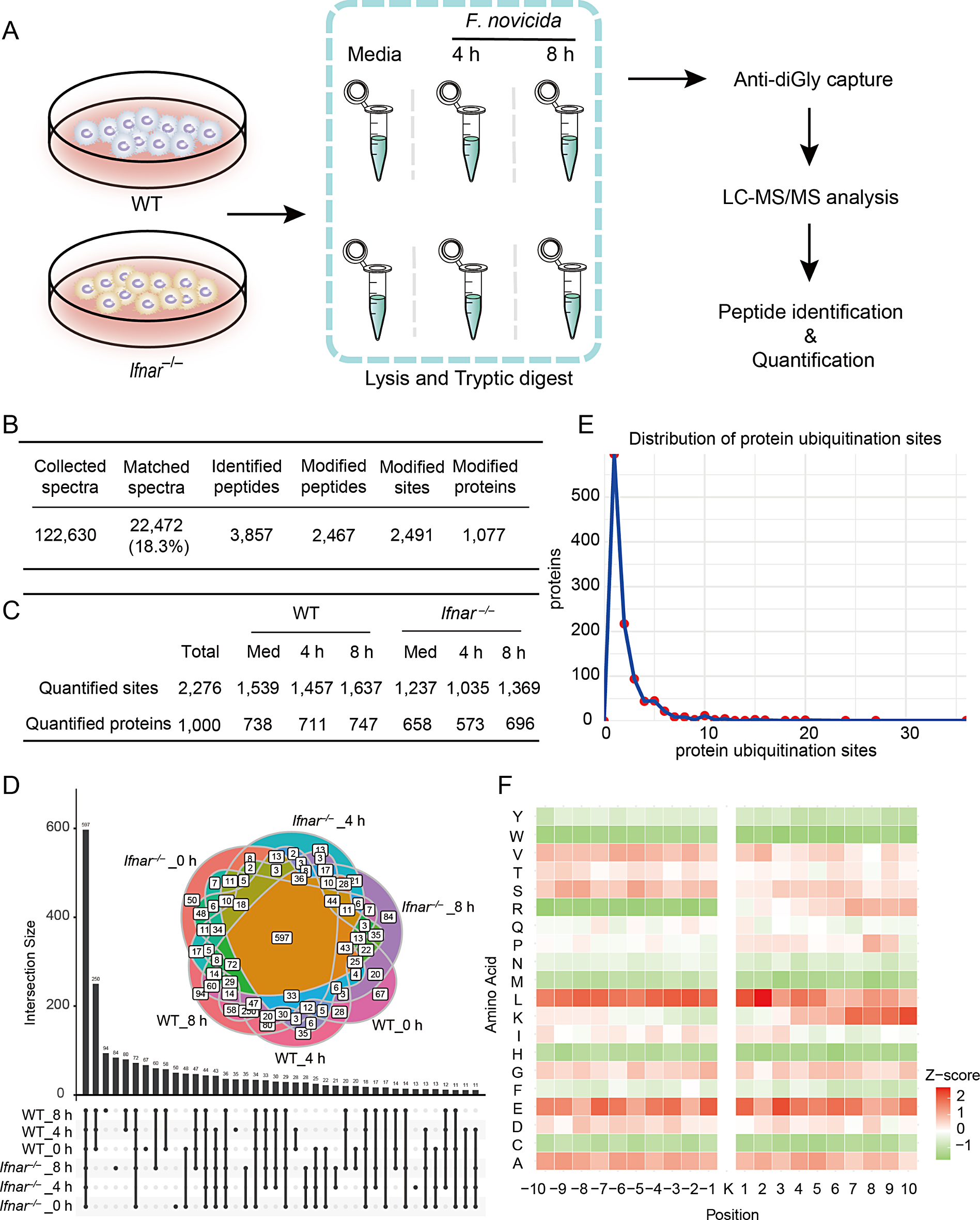
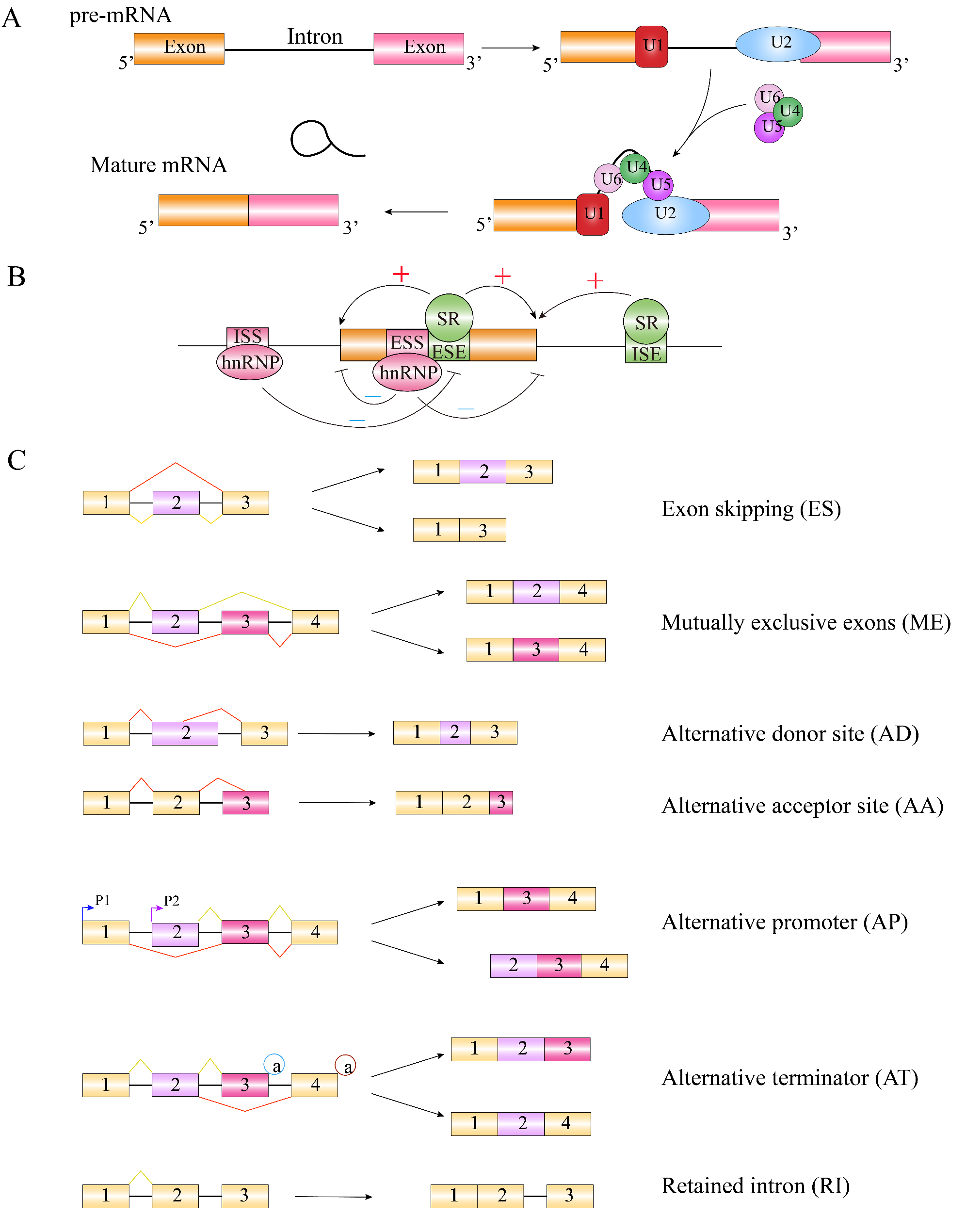
Remember me


Ubiquitination is a reversible protein post-translational modification by covalently conjugating ubiquitin, a small 76-amino acid protein, to lysine residues in the target protein [1]. Like methylation and acetylation, ubiquitination is also coded to regulate cellular functions through the coordinated actions of enzymes, including “writers” for conjugating ubiquitin to the substrate, “readers” for ubiquitin recognition and function execution, and “erasers” for ubiquitin removal [2, 3]. The ubiquitin writer is a sequential three-step enzymatic reaction system that is mediated by ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). There are 2 E1 enzymes, 30–50 E2 enzymes, and over 600 human E3 ligases in the human genome [4]. The diversity of substrate-binding E3 ligases codes various types of polyubiquitination featured with distinct polyubiquitin linkages. Each ubiquitin code, such as the unique polyubiquitin linkage, is recognized by a different reader, leading to a specific fate of ubiquitinated proteins and distinct functional consequences. For example, lysine 48 (K48)-linked polyubiquitin is recognized by 26S proteasome, resulting in protein degradation. Similarly, the conjugated polyubiquitin is removed by erasers, a group of enzymes known as deubiquitinases (DUBs), providing the recycling of ubiquitin into the cytosolic pool and a counterbalance to the ubiquitin-mediated signaling pathways.
Ubiquitination is critical for cell homeostasis and plays a vital role in many physiological processes, including protein degradation, signal transduction, DNA repair, cell proliferation, and immune response. Mounting evidence highlights the disruption of ubiquitin code in aberrant cell signaling and disease development and progression, including various types of cancers. For example, alterations in the activity of many E3 ligases and DUBs are significantly associated with the etiology of human malignancies [5]. There are many excellent reviews on the role of well-known ubiquitin codes, like K48- and K63-linked polyubiquitin, in cancers [5,6,7]. Recently, a new ubiquitin code, the N-terminally Methionine 1-linked linear polyubiquitin, has been found. Although linear ubiquitin is much less abundant than other types of ubiquitin chains, recent studies have found that they play pivotal roles in tumorigenesis and cancer pathogenesis. However, there is no comprehensive review of the emerging role of linear ubiquitination in cancer. In this review, we focus on the role of linear ubiquitin in cancers, delineate linear ubiquitin-mediated signaling pathways, and discuss current therapeutic approaches that target linear ubiquitination for cancer therapy.
Linear ubiquitination process and regulationMost types of polyubiquitin linkage are determined by the covalent bond formed between the C-terminal glycine residue of one ubiquitin molecule and a lysine residue on the previous ubiquitin molecule, such as the K48- and K63-linked ubiquitination. However, linear ubiquitination is a type of non-canonical linkage characterized by the head-to-tail linkage of ubiquitin molecules via the C-terminal carboxyl group of the donor ubiquitin and the N-terminal methionine of the acceptor ubiquitin. This results in the formation of a peptide bond in contrast to isopeptide formation via the linkage to the epsilon amino group of a lysine residue. This section discusses the writer, readers, erasers, regulators, and substrates for linear ubiquitination (Fig. 1).
Fig. 1
Schematics of the writer, reader, eraser, and regulator for linear ubiquitination. Linear ubiquitin chains are assembled by the E3 ligase LUBAC along with the E2 UBE2L3. LUBAC comprises HOIP, HOIL-1, and SHARPIN, which serves as the linear ubiquitin writer. Several proteins decode linear ubiquitin by specifically binding to translate into a cellular effect (readers). Two deubiquitinases disassemble linear ubiquitin chains as the erasers. A20, TNF-inducible protein A20; CYLD, cylindromatosis; HOIL-1, heme-oxidized IRP2 ubiquitin ligase 1L; HOIP, HOIL-1-interacting protein; OTULIN, OTU domain-containing deubiquitinase with linear linkage specificity; SHARPIN, SHANK-associated RH domain-interacting protein; UBAN, ubiquitin-binding domain in ABIN proteins and NEMO; UBE2L3, ubiquitin-conjugating enzyme E2 L3. The figure was created with BioRender.com
Writer: the LUBAC complexThe writer for linear ubiquitin chains is the E3 ligase complex, the linear ubiquitin chain assembly complex (LUBAC) [2], along with the ubiquitin-conjugating enzyme E2 L3 (UBE2L3) [8]. LUBAC is a ~ 600 kDa complex consisting of three subunits, including two RING-between-RING (RBR)-type ubiquitin ligases, heme-oxidized IRP2 ubiquitin ligase 1L (HOIL-1L, also known as RBCK1) [9] and HOIL-1-interacting protein (HOIP, also known as RNF31) [10], and one adaptor protein SHANK-associated RH domain-interacting protein (SHARPIN) [11,12,13] (Fig. 2). Although HOIP and HOIL-1 are both E3 ligases, HOIP is the catalytically active component of LUBAC and the only E3 ubiquitin ligase that can assemble linear ubiquitin. HOIP recognizes ubiquitin-bound E2 at its RING1 domain and transfers ubiquitin from E2 to the active Cys885 in the RING2 domain via a thioester-linkage. Then, the C-terminal linear ubiquitin chain determining domain (LDD) facilitates the transfer of ubiquitin to the acceptor ubiquitin to form a linear linkage [14,15,16,17]. Interestingly, the catalytic activity of HOIP is autoinhibited by its N-terminal domain. The binding of the ubiquitin-like (UBL) domain of HOIL-1L and SHARPIN to the ubiquitin-associated domain (UBA) of HOIP releases HOIP from autoinhibition [14,15,16, 18, 19], suggesting that the integrity of LUBAC is critical for its activity. Furthermore, the E3 activity of HOIL-1L also regulates LUBAC activity. HOIL-1L conjugates monoubiquitin onto all LUBAC subunits, followed by HOIP-mediated conjugation of linear chains onto mono-ubiquitin, which attenuates the functions of LUBAC [20]. Overall, LUBAC is the sole E3 ligase complex that facilitates linear ubiquitination, which depends on the integrity of its three essential components.
Fig. 2
Domains and post-translational modification sites of the LUBAC subunits, OTULIN, and CYLD. CAP-Gly, cytoskeleton-associated protein glycine-rich; IBR, in-between RING; LDD, linear ubiquitin chain determining domain; LTM, LUBAC-tethering motif; OTU, ovarian tumor; PH, Pleckstrin-homology; PIM, PUB-interacting motif; PR, proline-rich region; PUB, PNGase/UBA or UBX; RING, really interesting new gene; UBL, ubiquitin-like; NZF, Npl4-type zinc finger; UBA, ubiquitin-associated; USP, ubiquitin-specific protease; ZF, zinc finger. The figure was created with BioRender.com
ReadersUbiquitin chains are recognized by readers through their ubiquitin-binding domains. These readers further decode each type of ubiquitin linkage to execute specific functions. The readers for linear ubiquitin are UBAN (UBD in ABIN proteins and NEMO) domain-containing proteins [21] and A20 (also known as TNFAIP3) [22, 23]. The UBAN domain binds linear polyubiquitin and is shared by several proteins involved in NF-κB signaling pathways, including NF-κB essential modulator (NEMO), the A20 binding and inhibitor of NF-κB 1 (ABIN-1), ABIN-2, ABIN-3, and Optineurin (OPTN) [21]. Among these readers, the mechanism of how NEMO decodes linear ubiquitin is extensively studied. The UBAN of NEMO has a high affinity with linear ubiquitin via a surface on the proximal ubiquitin moiety and the canonical Ile44 surface on the distal one [24, 25]. The linear ubiquitin chain binding of NEMO recruits the IκB kinase (IKK) complex to the activation signalosome platform and induces IKK oligomerization and subsequent NF-κB activation [26]. A20 has a dual function as both DUB and E3 ligase due to the N-terminal OTU domain and a C-terminal zinc finger domain [27]. However, the DUB activity of A20 is not required for the NF-κB suppression. Instead, the interaction with linear ubiquitin through the ZF7 domain of A20 is crucial for NF-κB suppression [22, 23]. Thus, A20 is classified as a linear ubiquitin reader but not an eraser. In addition, HOIL-1L, the OTU domain-containing deubiquitinase with linear linkage specificity (OTULIN, also known as FAM105B or Gumby), and cylindromatosis (CYLD) are linear ubiquitin-binding proteins; however, their binding contributes to their roles in linear ubiquitin chain generation and removal, as discussed elsewhere. Collectively, NEMO and A20 are two extensively researched linear ubiquitin readers. Further exploration of proteins that bind to linear ubiquitin will likely uncover additional readers.
Erasers: OTULIN and CYLDThe deubiquitinases, OTULIN and CYLD, bind to linear ubiquitin chains through their catalytic domains and hydrolyze linear polyubiquitin [28,29,30,31,32,33,34] (Fig. 2). OTULIN exclusively disassembles linear ubiquitin chains [28,29,30] while CYLD hydrolyzes both K63-linked and linear ubiquitin chains [32,33,34]. Furthermore, OTULIN but not CYLD prevents LUBAC from auto-ubiquitination [28, 30, 34, 35]. In addition, OTULIN knockout induces a strong increase in the abundance of linear ubiquitin [29, 36], which is not observed in CYLD-deficient cells [37], suggesting that these two erasers have distinct functions in the removal and regulation of linear polyubiquitin. A recent genetic study found that OTULIN promotes rather than counteracts LUBAC activity by preventing its auto-ubiquitination with linear polyubiquitin [35]. Although OTULIN and CYLD both are linear ubiquitin erasers, they have distinct phenotypes in vivo. While CYLD knockout mice show no major defects and are born at expected ratios [38], OTULIN null mutant mice (known as Gumby mice) are embryonic lethal [29]. These differences likely stem from their unique noncatalytic functions and interacting partners, which warrants further investigation in the future.
RegulatorsRecent studies show that the linear ubiquitin writer and erasers are tightly regulated by proteases and post-translational modifications, adding complexity to the different layers of linear ubiquitination. First, the two components of LUBAC, HOIP, and HOIL-1L, are cleaved by proteases. HOIP is cleaved at aspartate 348 (D348), D387, and D390 by caspases 3 and 6 during apoptosis [39, 40] and caspase 8 in the TNF-related apoptosis-inducing ligand (TRAIL)-induced cell death [41]. HOIL-1L is cleaved by paracaspase, mucosa-associated lymphoid tissue lymphoma translocation gene 1 (MALT1) [42,43,44]. MALT1 cleaves HOIL-1L between arginine 165 (R165) and glycine 166 (G166), resulting in the removal of the HOIL-1L RBR domain [44]. The RBR domain of HOIL-1L augments LUBAC activity, which is associated with the pathogenesis of B-cell-like diffuse large B-cell lymphoma [44, 45].
In addition to protease cleavage, HOIP is regulated by ubiquitination and phosphorylation. HOIP is ubiquitinated of K1056 at the carboxyl terminus by unknown E3 ligase(s) [46]. This ubiquitination dynamically alters HOIP conformation, resulting in the suppression of its catalytic activity. HOIP is phosphorylated at S1066 by the mammalian ste20-like kinase 1 (MST1), which attenuates the E3 ligase activity of LUBAC [47]. Similarly, SHARPIN is also a phosphorylated protein. SHARPIN is constitutively phosphorylated on serine 165 (S165) by an unknown kinase in lymphoblastoid cells. Moreover, this phosphorylation can be further enhanced by ERK1/2 kinases upon T cell receptor (TCR) engagement [48]. The S165A mutation of SHARPIN impairs the linear ubiquitination of NEMO and hinders NF-κB activation, suggesting that SHARPIN controls optimal activation of NF-κB response to both TCR and TNFα stimulation.
Like the ubiquitin writer, CYLD and OTULIN are also regulated by post-translational modifications. OTULIN activity is regulated by two post-translational modifications, phosphorylation and ubiquitination. OTULIN is phosphorylated at tyrosine 56 (Y56) located in the PUB domain-interacting motif (PIM). The PIM is responsible for binding to the N-terminal PUB domain of HOIP; however, the Y56 phosphorylation abrogates OTULIN-HOIP interaction [49,50,51]. Interestingly, OTULIN is hyper-phosphorylated at Y56 during necroptosis and counteracted by the dual specificity protein phosphatase 14 (DUSP14) [52]. Recently, Wang et al. reported that the ABL1 tyrosine kinase phosphorylates OTULIN at Y56 and promotes genotoxic Wnt/β-catenin activation to enhance drug resistance in breast cancers [53]. In addition, our lab recently found that OTULIN is subject to non-proteolytic ubiquitination upon TNFα stimulation [54]. We found that the E3 ligase tripartite motif-containing protein 32 (TRIM32) interacts with the OTU domain of OTULIN and conjugates non-proteolytic K63-linked polyubiquitin at K64 and K66 in the vicinity of the PIM domain. The polyubiquitin disrupts HOIP-OTULIN interaction, thereby enhancing TNFα-induced NF-κB activation [54].
CYLD protein stability is regulated by two different modifications. First, CYLD is subject to K48-linked ubiquitination and subsequent protein degradation. CYLD is ubiquitinated at K338 and K530 by the E3 ligase mind bomb homolog 2 (MIB2), which leads to CYLD proteasomal degradation and NF-κB activation [55]. CYLD is also targeted by another E3 ligase SCFβ−TRCP for degradation [56]. Second, CYLD is cleaved by proteases. It has been shown that caspase 8 (CASP8) cleaves CYLD at D215 in response to TNF and TLR stimulation [57, 58]. By contrast, upon TCR stimulation, the paracaspase MALT1 cleaves CYLD at arginine 324 (R324) [59]. In addition, CYLD is phosphorylated at the serine cluster between amino acids 418 and 444 by IKKα/β in response to TNF stimulation, which prevents CYLD from deubiquitinating the TNF receptor-associated factor 2 (TRAF2) and promotes TNF-induced gene expression [60]. Another IKK family member IKKɛ phosphorylates CYLD at S418, leading to decreased DUB activity [61]. By contrast, a recent study showed that IKK β phosphorylates CYLD at S569 to increase its DUB activity [62]. More importantly, S568 phosphorylation, in concert with S418 phosphorylation, switches CYLD’s DUB activity toward K63-linked polyubiquitin [63]. Furthermore, spermatogenesis-associated protein 2 (SPATA2) bridges CYLD to HOIP by binding to CYLD and HOIP via its PUB domain and PIM, respectively [64,65,66,67]. The SPATA2-mediated CYLD-HOIP interaction is essential for CYLD activity to linear ubiquitination and NF-κB activity [64,65,66,67].
Lastly, linear polyubiquitin itself is also regulated by post-translation modification. Wu et al. found that TNF induces the recruitment of LUBAC and the assembly of linear ubiquitin chains at mitochondria [68]. LUBAC stabilizes the PTEN-induced kinase 1 (PINK1) at the outer mitochondrial membrane by linear ubiquitination. PINK1 further phosphorylates linear ubiquitin at S65, which counteracts their OTULIN-mediated hydrolysis and augments NF-κB signaling [
Comments (0)