Study population and sample collection protocols
Umbilical cord blood samples (10 from normal pregnancies and 10 from PE) were collected at the Department of Obstetrics of Shandong Provincial Hospital after approval by the Ethics Review Committee of Shandong Provincial Hospital. All patients provided preoperative informed consent. After the myometrium was incised and the placenta was delivered, whole blood was collected by aseptic acupuncture before caesarean section via venepuncture in anticoagulant EDTA-K2 (ethylenediaminetetraacetic acid–K2) and centrifuged at 4000 × g for 15 min. Normal control pregnancies were defined as having no pregnancy complications (n = 10, 38.5 ± 0.5 weeks). The PE group was defined as maternal blood pressure (≥ 140/90 mmHg) and urinary protein (≥ 1 +) at 20 weeks of gestation (n = 10, 37 ± 1.9 weeks), and those with diabetes and other pregnancy complications were excluded [16, 44]. More clinical information about the sample can be found in Table 1.
Table 1 Demographic and clinical characteristics of the study populationCell culture
HTR8/SVneo cells (human first-trimester extravilloustrophoblast cells) were purchased from ATCC. Non-treated (NO) cells were routinely cultured in RPMI 1640 (Gibco) supplemented with 5% exosome-free foetal bovine serum (Gibco) and 1% penicillin/streptomycin at 37 °C and 5% CO2. Hypoxia/reoxygenation (H/R) was performed as previously described [45]. The trophoblast cells were cultured in two cycles: the first was in a hypoxic environment in a tri-gas cell culture incubator flushed with 2% O2 for 8 h, and this was followed by reoxygenation in a standard incubator with 20% O2 for 16 h.
Human glomerular endothelial cells (HGECs) were purchased from ATCC. The cells were routinely cultured in DMEM (Gibco) with 10% exosome-free FBS and 1% penicillin/streptomycin at 37 °C and 5% CO2.
ELISA
ELISA for human HIF-1α were performed according to the manufacturer’s instructions. In brief, HTR8/SVneo cell lysates buffer (NO and H/R) were collected, and the precipitate was removed by centrifugation for 10 min at 4 °C incubated in a 96-well plate precoated with capture antibodies. Samples were added to the plate. Wells were washed 5 times and incubated with a secondary antibody conjugated to horseradish peroxidase. Then the substrate solution was added, and the optical density was determined at 450 nm. The protein levels were calculated using a standard curve derived from known concentrations of the respective recombinant proteins.
Exosomes isolation
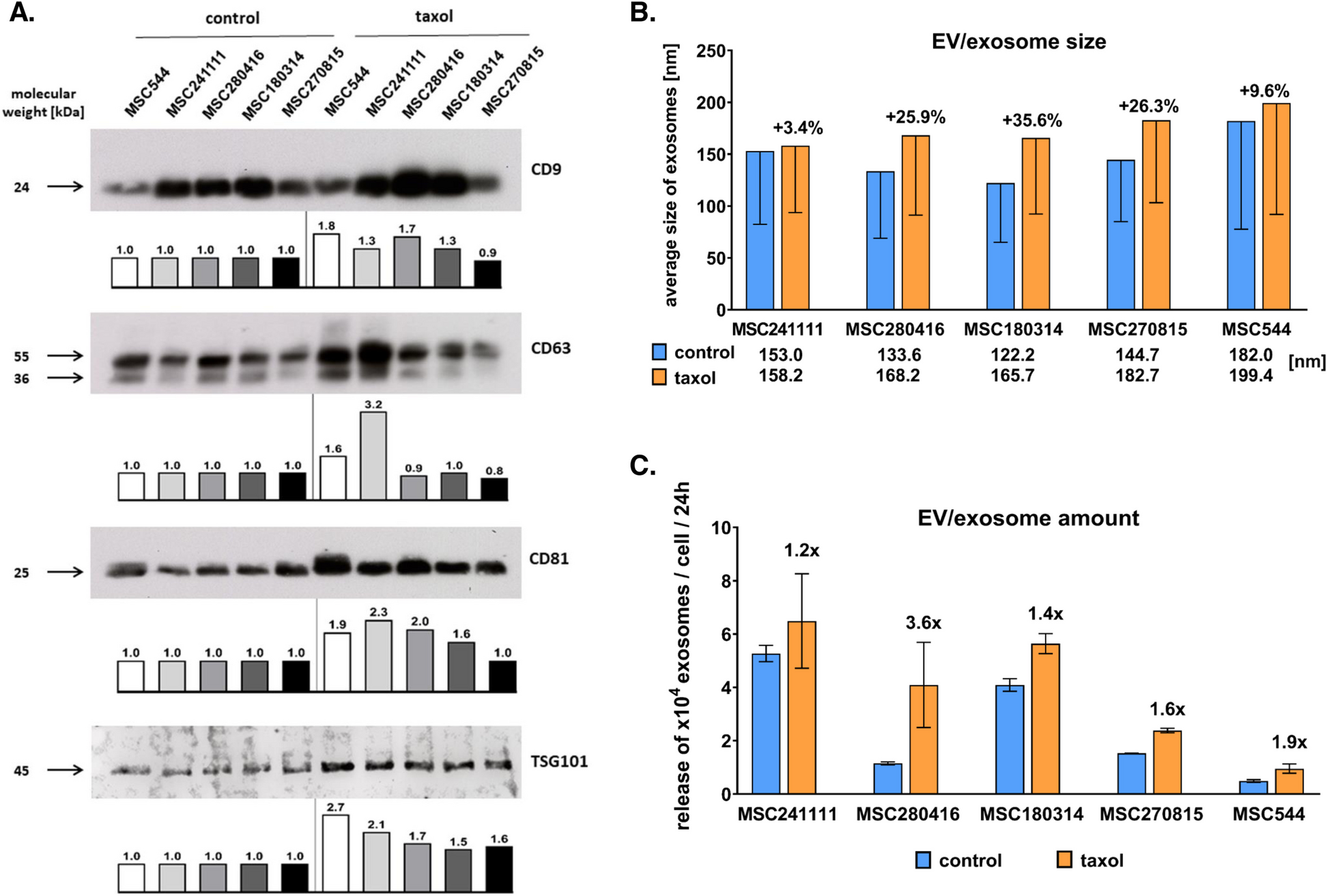
Trophoblast-derived exosomes from HTR8/SVneo cells were extracted by ultracentrifugation [46, 47]. In simple terms, the culture medium was centrifuged at 4 °C at 500 × g, 2000 × g and 12,000 × g for 10 min, 30 min and 45 min, respectively, to remove whole cells and debris. The resulting supernatant was sterilized by means of a 0.22-μm filter and centrifuged at 120,000 × g (Hitachi CP100MX) for 70 min. The particles were resuspended in PBS and centrifuged after washing (120 000 × g, 70 min). Finally, the exosomes were suspended in 200 µl of PBS and placed in a -80 °C freezer for subsequent experiments. The concentration of exosomes was measured with a BCA protein analysis kit (Solarbio, Beijing, China).
Placenta-derived exosomes from cord blood were extracted by a series of sucrose density gradient centrifugations and ultracentrifugation [42, 43]. In simple terms, 20 ml of cord blood was centrifuged at 4 °C at 3000 × g for 20 min to produce 8 ml plasma. Next, an equal volume of 1 × PBS was added and centrifuged at 4 °C at 500 × g, 2000 × g and 12,000 × g for 15 min, 30 min and 45 min, respectively. The supernatant was taken, and the precipitate was discarded to remove cells, cell fragments, large vesicles and small vesicles. The supernatant was filtered through a 0.22-µm membrane, and the supernatant mixture was suspended in 0.25 M-sucrose. The suspension was then stratified on a sucrose density gradient, centrifuged at 4 °C at 200 000 × g for 16 h, and divided into six fractions: F1, 1.03; F2, 1.06; F3, 1.09; F4, 1.11; F5, 1.14; and F6, 1.18 g/ml. The fractions were centrifuged at 4 °C and 120,000 × g for 2 h. Then, in order to enrich placenta-derived exosomes in umbilical cord blood, we collected F2 and F3 exosomes and suspended them in 200 µl PBS for subsequent experiments. The concentration of exosomes was measured with a BCA protein analysis kit (Solarbio, Beijing, China).
Transmission electron microscopy
Exosomes (10 μl) were added to copper wire for precipitation for 1 min, and the floating liquid was absorbed on filter paper. Ten microlitres of phosphotungstic acid was added to the copper wire for precipitation for 1 min, and the floating liquid was absorbed by filter paper. After drying for several minutes at room temperature, TEM imaging results were obtained at 100 kV (Xiuyue Biol, Jinan, China).
Nanoparticle tracking analysis
Ten microlitres of exosome sample was removed and diluted to 30 μl. The instrument performance test was first performed with the standard product. After passing the test, the exosome sample was loaded. The particle size and concentration of exosomes detected by the instrument were obtained after the samples were tested (Xiuyue Biol, Jinan, China).
Exosome labelling
The exosomes were labelled with the fluorescent dye PKH67 (green) (PKH67; Sigma) and incubated for 20 min according to the manufacturer’s protocol. The 17-labelled excised suspensions were filtered with a 100-kDa intercepted hollow fibre membrane to remove excess dye. HGECs were inoculated in 96-well plates and incubated with double-labelled exosomes (100 μg/ml) for 24 h. Kidney explants were inoculated in 24-well plates and incubated with double-labelled exosomes (200 μg/ml) for 96 h. HGECs were fixed in 4% paraformaldehyde for 10 min, 100 μl of diluted phalloidin was added to each well and incubated at room temperature for 30 min, and DAPI staining solution was added for 5 min. Then, labelled cells were prepared and observed under ImageXpress Microconfocal with MetaXpress software (overall magnification 100X).
To fluorescently label exosomes for in vivo imaging, we resuspended the pellet centrifuged at 100,000 × g for 2 h in 7.0 ml of 7.5 µM DiR (Life Technologies, Carlsbad, CA, USA) in PBS. After mixing, the exosomes were incubated in the DiR/PBS solution for 15 min at room temperature in the dark and then ultracentrifuged at 100,000 × g for 1 h. The final pellet was resuspended in 50 µl of PBS and stored at − 80 °C.
EdU staining
EdU staining was performed according to the manufacturer's instructions (Beyotime, Shanghai, China). Briefly, HGECs (5 × 103 cells/well) were incubated with different exosomes (NO-exo, H/R-exo, N-exo, PE-exo, 100 μg/ml) for 24 h. EdU (10 μM/well) was then added to the medium and incubated for 4 h. After labelling, the cells were washed three times with PBS and then fixed with 4% formaldehyde. After incubation with glycine, the cells were washed with PBS containing 0.5% Triton X-100. After the nuclei were stained with DAPI, cell proliferation was observed by ImageXpress Micro Confocal with MetaXpress software (overall magnification 200X).
Tube formation assay
To quantitatively determine the ability of HGECs to generate blood vessels in vitro, we applied substrate glue to the bottom of 96-well plates. HGECs (1 × 104 cells/well) were added to serum-free endothelial cell culture medium after Matrigel mix (BD Bioscience) coagulation, and the cells were incubated with different exosomes (NO-exo, H/R-exo, N-exo, PE-exo, 100 μg/ml) for 6 h. Finally, the cells were photographed with an inverted microscope (Thermo) for analysis; ImageJ was used to quantify tube formation.
Cell migration assay
The migration of HGECs was measured using Transwell inserts (Corning, USA) with 8 μm polycarbonate membranes. The process was as follows: 200 µl of serum-free medium was added to the upper chamber, and 650 µl of complete medium containing 5% serum was added to the lower chamber. HGECs (1 × 104 cells/well) were incubated in the upper chamber with exosomes from the different groups (NO-exo, H/R-exo, N-exo, PE-exo, 100 μg/ml). After 24 h of culture, the cells that migrated to the lower chamber were stained with crystal violet. Finally, an inverted microscope (Olympus, Tokyo, Japan) was used at a magnification of 200X to count the average number of migrated cells.
Cell permeability assay
The flow of Evans blue bound to albumin across the monolayer of a functional artificial liver was measured by spectrophotometry using a modified two-compartment model that was described previously for quantitative permeability [48]. In brief, HGECs were plated (5 × 104 cells/well) in a Transwell tube with diameters of 0.4 μm and 6.5 mm for 3 days. Confluent monolayers were incubated with different exosomes (NO-exo, H/R-exo, N-exo, PE-exo, 100 μg/ml) for 24 h. The inserts were washed with PBS (pH 7.4), and then, 0.5 ml (0.67 mg/ml) of Evans blue BSA (4%) diluent was added to the medium. Fresh medium was added to the lower chamber, and Evans blue BSA was added to the upper chamber. After 10 min, the optical density at 650 nm in the lower chamber was measured. The experiment was repeated several times in triplicate.
Animal study
C57 male and female mice (6–8 weeks) were purchased from Jinan Pengyue Experimental Animal Breeding Company and kept in a temperature-controlled room at 24 °C with a light/dark cycle of 12:12 h and free access to food and water. This project was implemented in accordance with the Animal Protocol procedures approved by the Department of Laboratory Animal Science, Shandong University Affiliated Provincial Hospital Animal Laboratory, and animals were processed in accordance with guidelines published in the National Institutes of Health under the guidance of the Animal and Institutional Animal Health and Use Committee. To evaluate the gestational age of mouse embryos consistently and accurately, we paired male and female C57 mice for one night. The first vaginal plug was found on embryonic day 0.5 (E0.5).
Foetal kidney culture in vitro
Foetal kidneys of E12.5 mice were isolated in vitro and randomly divided into different group as described previously [49,50,51]. In brief, the foetal kidney was transferred to a Transwell semipermeable membrane, and 500 µl of DMEM/F12 culture medium was added to each well and incubated in an incubator. On the second day, the old culture medium was discarded, and NO-exo, H/R-exo, N-exo and PE-exo were added (200 µg/ml, 3 multiple wells/group). The kidney was cultured in the incubator for another 4 days, and the growth of the kidney was observed by taking pictures with an inverted microscope. The ureteral buds (E-cadherin, green) and glomerulus (WT-1, red) of foetal kidneys cultured for 5 days were stained by immunofluorescence and photographed by a laser confocal microscope to observe the structural changes of foetal kidney development. Foetal kidney protein and RNA were extracted, and the growth and development of the foetal kidney were quantitatively analysed by Western blot analysis and qPCR.
Amniotic cavity injection
For confirmation that exosomes could enter the foetus, E14.5 mice were continuously anaesthetized with isoflurane, and the mice received minor open surgery and intrauterine injection of DIR-labelled exosomes as described previously [52]. Twenty-four hours later, the animal’s lateral abdomen was captured by an in vivo imaging system (Tanon ABL X6, China). After in vivo imaging, the animals were sacrificed by inhaling carbon dioxide in accordance with IACUC and American Veterinary Medical Association guidelines.
Pregnant mice with similar body weights on the same day were randomly divided into different group. NO-exo, HR-exo, N-exo and PE-exo (500 µg/ml) were injected into the amniotic cavity of pregnant mice on E14.5. The foetal mice were removed by caesarean section on E18.5, and the kidneys were separated. PAS staining and immunohistochemistry staining were performed to observe the structural changes during foetal kidney development. Foetal kidney protein and RNA were extracted, and the growth and development of the foetal kidney were quantitatively analysed by Western blot analysis, IHC and qRT-PCR.
Western blot analysis
Total protein was extracted from HTR8/SVneo cells, exosomes or HGECs, explants, and kidneys treated with exosomes. The total protein concentration was measured with a BCA kit. Next, the proteins were separated by gel electrophoresis and then transferred to polyvinylidene fluoride membranes using electrical blotting. Ten micrograms of total protein from each sample was used for HIF1-α, Golgi marker, exosome-specific antibodies and junction protein-specific antibodies. The antibody HIF1-α (1:1000; Abcam). The Golgi marker GM130(1:1000; Abcam). The exosome-specific antibodies included CD63 (1:1000; Abcam) and TSG101 (1:1000; Abcam); the placenta-specific antibody was PLAP (1:1000; Abcam). The junction protein-specific antibodies included VE-cadherin (1:1000; CST) and Occludin (1:1000; Abcam) and the housekeeping antibodies included β-actin (1:1000; CST). The antibodies were incubated with the blots overnight at 4 °C. The blots were then incubated with HRP-conjugated goat anti-rabbit secondary antibodies (Proteintech, Rosemont, IL) at room temperature for 1 h. The immunofluorescence bands were detected with a kit (Merck Millipore, Burlington, MA), and the intensity of the bands was quantified by an Amersham Imager 600 Imaging System (GE Healthcare, Chicago, IL).
Quantitative RT-PCR (qRT-PCR) analysis
HGECs, explants, and kidneys were treated with different exosomes (NO-exo, H/R-exo, N-exo, PE-exo). A StepOnePlus real-time quantitative PCR system (Invitrogen, CA, USA) was used to measure mRNA. The VE-cadherin primers (5´-ACGACAACTGGCCTGTGTTCAC and 3´-TG-CATCCACTGCTGTCACAGAG) yielded a 101-base pair fragment. The Occludin primers (5´-ACCCCCATCTGACTATGTGGAA and 3´-AGGAACCGGCGTGGATTTA) yielded a 115-base pair fragment. The Gapdh primers (5´-AGATCCCTCCAAAATCAAGTGG and 3´-GGCAGAGATGATGACCCTTTT) yielded a 130-base pair fragment. SYBR® Premix Ex Taq™ (Accurate Biotechnology, Hunan, China) was used for amplification, and gene expression was calculated with the 2 − ΔΔCT method (with Gapdh as an internal reference).
Immunofluorescence, periodic acid-schiff, and immunohistochemistry
Immunofluorescence was used to detect the expression of ureteral buds and glomeruli in explants. The tissues were fixed with 4% paraformaldehyde for 48 h and treated as usual. For paraffin-embedded tissue, sections of 3–5 μm thick paraffin-embedded tissue were cut and dewaxed with xylene, dehydrated with ethanol, and stained with periodic acid-schiff (PAS). The expression of VE-cadherin and Occludin in tissue sections was detected by immunohistochemistry (IHC).
Statistical tests
The data are expressed as the means ± SDs of three independent experiments. ImageJ software was used for data analysis, and GraphPad Prism (ver. 7; GraphPad Software, Inc., La Jolla, CA) was used for statistical analysis. Student’s t tests were used to analyse differences between the two groups. P < 0.05 was considered statistically significant (ns, P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).


Comments (0)