
Remember me
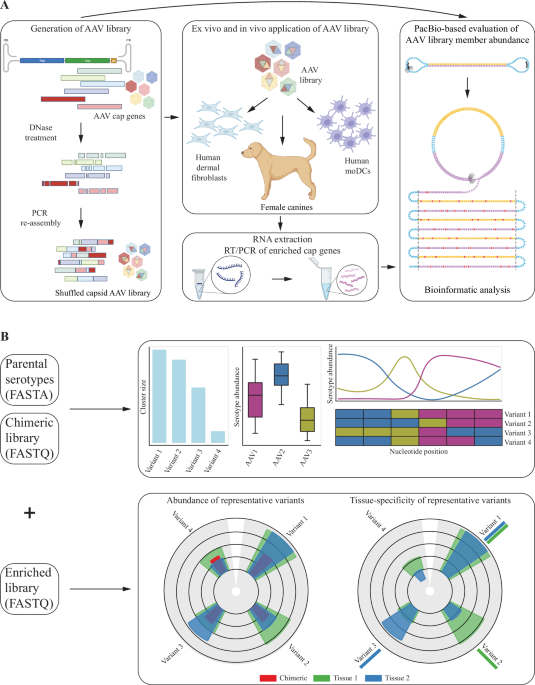
The hafoe program takes as input two files: a FASTQ or a CSV formatted file containing PacBio circular consensus sequences (CCS) and abundances of the chimeric DNA sequences and a FASTA file containing the cap gene sequences of the parental AAV serotypes. Assembled AAV sequences generated from other long-read sequencing platforms, such as Oxford Nanopore, can also be supplied in the same formats. Optionally, it can also take one or more additional files of FASTQ format containing the sequences of selected variants in the enriched libraries. hafoe can take as input multiple input libraries describing the enriched chimeric variants in different target tissues. In addition to text files, hafoe also produces interactive graphics in HMTL reports (Fig. 1B).
Fig. 1: Schematic overview of experimental design and workflow of hafoe.
A Experimental design for generating chimeric AAV capsid library from parental AAV serotypes, followed by enrichment in DCs and HDFs, total RNA extraction, and PacBio sequencing. B hafoe takes as input parental AAV serotypes and libraries of chimeric variants before and/or after enrichment in target tissues. hafoe produces interactive graphics representing the identified clusters of similar chimeric vectors, showing the prevalence of each wild-type vector and serotype composition of chimeric representatives. It also shows the enrichment of representative sequences in target tissues and the tissue-specificity of each vector.
PreprocessingBoth chimeric and enriched library files are filtered by sequence length and the open reading frame (ORF) boundaries. The ORFs are identified as the longest sequence regions starting from the start codon ATG and ending with either of the three stop codons (TAA, TAG, TGA) in six possible reading frames. Only variants with ORF size greater than 1.8 kb and total length less than 3 kb are selected for downstream analysis. To reduce sequence redundancy in the resulting chimeric library that arises as a result of point mutations or sequencing errors, we cluster the library members using the CD-HIT-EST program of the CD-HIT package version v4.8.1 [25] (Fig. 2A). The program performs clustering by first sorting the input sequences by length; the longest one becomes the representative of the first cluster, the remaining sequences are compared with it and if the similarity is above the selected threshold (-c 0.9), the sequences are grouped into that cluster; otherwise, the dissimilar sequence becomes a representative of a new cluster. For each comparison, CD-HIT applies a minimum number (-n 9) of identical short substrings of a given length shared by two sequences [25]. If the number of such substrings is below the set threshold, an actual sequence alignment is performed. As a similarity measure, the local sequence identity (-G 0) between two sequences is calculated by dividing the number of identical bases in alignment by the alignment length, where the alignment coverage for the longer sequence is specified as (-aL 0.9). As per our specifications, each sequence is assigned to the cluster of the highest similarity (-g 1). After clustering, hafoe re-assigns cluster representatives as the most abundant sequences in the chimeric library.
Fig. 2: hafoe’s neighbor-aware serotype identification algorithm.
A Chimeric library cluster size distribution. hafoe clusters the chimeric library sequences based on sequence identity and uses the representative variants of the clusters in subsequent analysis. B To identify the variant composition in terms of the AAV serotypes, hafoe first performs variant decomposition: chopping the variant into overlapping fragments of fixed size, aligning the fragments against AAV serotypes, and storing the alignment results in a list. C hafoe then performs neighbor-aware serotype identification with the following steps: for each position covered by fragments with multiple assignments Step 1) perform quality filtering (see methods), Step 2), identify the set of serotypes shared with its left and right neighbors if those exist (left_intersect and right_intersect), and update the position accordingly, Steps 3) annotate the unresolved positions as multimappers (e.g. with 17).
Neighbor-aware serotype identificationNeighbor-aware serotype identification is a method we have designed to describe the variant sequences and identify the variants’ compositions in terms of parental AAV serotypes. For each input sequence, we output a list of parental serotypes from which their different fragments potentially originate.
First, the variants’ sequences are chopped into overlapping fragments of equal size. By default, the fragment length is set to 100 bp, and the length of the region between the starting positions of two consecutive fragments is set to 10 bp. These default values are chosen based on the most optimal performance during our validation experiments with in silico datasets (see the text below and Supplementary Fig. 1). The fragments are then aligned to AAV serotype genomes using the Bowtie 2 program version v2.4.2 [26] (with options -a --no-unal -L 30 -D 2). The bowtie2 command is used with increased seed length (-L 30) and reduced number of seed extension failed attempts (-D 2) options to perform a strict alignment. For each variant, the serotypes its fragments align to are stored in a list (Fig. 2B).
This list is then used for neighbor-aware serotype identification, which assigns the most probable serotype to each fragment based on its neighborhood, assuming that neighboring fragments are more probable to originate from the same serotype (Fig. 2C). The workflow proceeds as follows:
1.For each nucleotide position of the variant sequence covered by fragments with multiple serotype assignments:
1.1we select the assignment with the maximum sum of alignment quality scores across corresponding fragments (see Supplementary Fig. 1 for the reasoning behind the selection of this metric). In case of ties, we keep all the assignments.
1.2we identify the sets of serotypes shared with its left and right neighbors (left_intersect and right_intersect in Fig. 2C) if those exist. If a position doesn’t have a left or right neighbor, we add one with an empty serotype set assignment.
1.3we assign to that position the union of serotypes in the left_intersect and the right_intersect sets if the union is not an empty set and leave the assignment unchanged otherwise.
2.If, after these steps, there are still unresolved positions with multiple serotype assignments, we annotate them as multi-mappers.
In the end, for each variant, hafoe outputs position-based assignments of parental serotypes or marks positions with multiple possible serotypes of origin.
With the default length of the region between the starting positions of two consecutive fragments (10 bp), hafoe detects crossover events at a resolution of 10 nt rather than determining the exact nucleotide position. To obtain nucleotide-resolved crossover positions the user can set this parameter to 1 bp.
Identification of enriched variantsAfter the assignment of serotype composition to each representative variant in the chimeric library, we wanted to explore which of those variants were successfully assembled, propagated, and enriched in the follow-up in vivo experiments in target tissues. For this purpose, hafoe uses the CD-HIT-EST-2D program of the CD-HIT package version v4.8.1 (with the options -g 1 -d 100 -c 0.95 -n 10 -G 0 -aL 0.95) to identify the sequences in the enriched library that are similar to the representative sequences in the chimeric library at 95% identity threshold. The total number of these sequences indicates the representative’s abundance in the enriched library. The fold change of a representative’s normalized abundance in the enriched library compared to its normalized abundance in the chimeric library was used as a measure of its enrichment. The representative variants with a log2 fold change greater than 1 were considered enriched, and those with a log2 fold change less than 1 were the reduced ones.
Library size normalizationThe percentages of each representative variant both in the chimeric and enriched libraries were used to normalize for library size. In the case of multiple enriched libraries provided as an input, hafoe performs normalization of variant abundances in chimeric and enriched libraries with the median-of-ratios method suitable for between-sample comparisons [27].
Analysis of peptide sequencesFor the analysis of these sequences, we employed the Xover tool’s algorithm (available at: http://qpmf.rx.umaryland.edu/xover.html) [20]. In brief, the Xover algorithm takes chimeric and parental protein sequences as input, performs multiple sequence alignment, and then, for each alignment position, compares the residue of the chimeric protein with the residues in parental sequences. For each position, the algorithm calculates the probabilities of the chimera’s residue originating from a given parent. Suppose there is a match between the chimera and a parental base at a given position. In that case, the algorithm evaluates the probability of the chimera’s residue originating from that parent as 1 divided by the total number of parental bases matching the chimera’s base at that position Otherwise, if there is no match, the probability is 0.
Software architecturehafoe is written in R 4.1.3 and Bash 5.1.4 and can be executed in Unix operating systems.
ORFik (v1.12.13), microseq (v2.1.4), and seqinr (v4.2.8) are the main R packages used in the program to filter the sequences, extract open reading frame (ORF) sequences, read and write files in FASTA and FASTQ formats, and read alignment files.
The Python bokeh package (v2.4.3) is used to visualize the obtained results and export them to HTML formatted report files. The general wrapper managing the overall workflow is written in Bash 5.1.4.
Accuracy of variant composition assessmentTo benchmark the different parameters of our algorithm, we have used different combinations of read length (the length of the chopped fragments from variant sequence), step size (the length of the region between the starting positions of two consecutive fragments), and filtering criterion (sum or average of alignment quality scores across all reads mapped to a position per serotype assignment (Fig. 2C, Step 1)) input parameters to run hafoe on five synthetic datasets. The accuracy of variant composition identification by hafoe was computed by dividing the total number of accurately described nucleotides in a sequence by the sequence length, and then averaging this value across all the sequences in the dataset (Supplementary Fig. 1).
Sequence conservation levels of AAV serotypesTo assess the conservation levels of parental AAV serotypes among groups of AAV variants, we first performed multiple sequence alignment of the variants of interest, using the Clustal Omega program (v1.2.4) (with the options specifying the output file format as –outfmt clu –force) [28]. Then a conservation score was derived for each position within the alignment by dividing the count of the most frequently occurring nucleotide at a given position by the total number of query sequences present in the multiple sequence alignment.
Experimental datasetsChimeric AAV capsid Library generation & virus productionTo generate the chimeric AAV capsid library, capsid (cap) genes from 16 wildtype AAV serotypes (AAV1-13, AAVrh8, AAVrh10, AAVrh32) were PCR-amplified and mixed equimolarly. These were fragmented using DNase I, yielding 200-400 base pair fragments, which were reassembled in primer-free cycles of denaturation, annealing, and extension (Fig. 1A). The shuffled chimeric AAV capsids were then PCR amplified using a BsaI overhang containing internal primer set, digested with BsaI, and ligated into an AAV2 Rep-containing ITR-backbone vector under a strong constitutively expressing ubiquitous EF1-alpha promoter. Ligation mixtures were then transformed into bacterial competent cells. The final chimeric library plasmids were then purified, and their size and diversity were validated using PacBio sequencing. For viral particle production of the shuffled chimeric AAV libraries, a triple-transfection protocol was used including an adenoviral helper plasmid (pHelper, Aldevron), Rep plasmid (pRep) and the above-made replication-competent shuffled AAV library using HEK293T cells by SignaGen (Fredrick, MD). Specifically, a 10-layer stack with 800ug of Helper plasmid, 800 ug of Rep plasmid, and 10ug of the ITR plasmid containing capsid library was used. Capsid sequences of successfully manufactured AAV variants were PCR amplified, recloned into the ITR vector and subjected to the second AAV production as described above. This final production yielded 3 mL of virus at 2.30E12 VG/mL. The production was scaled up to obtain 10 ml of 5E13vg/ml for the animal experiments.
Enrichment in dendritic cells and human fibroblastsPeripheral blood samples from healthy young adults were collected after obtaining written informed consent per protocol approved by the Institutional Review Board of Boston Children’s Hospital. De-identified, heparinized blood samples were processed within 2–4 h to isolate mononuclear (PBMCs) from the whole blood using Ficoll gradient, followed by monocyte purification using CD14+ beads (Miltenyi, 130-042-401) according to manufacturer’s protocol. Monocytes were cultured in Cellgro DC medium (Cellgenix GMBH, 20801-0500) with 250 IU/mL IL-4 ((Miltenyi, 130-093-922), and 800 IU/mL GM-CSF (Miltenyi, 130-093-866), and incubated in a 6-well plate at 37 °C and 5% CO2 for 5 days to generate Mo-DCs. On day 5, 2 × 106 non-adherent Mo-DCs were transferred to a new 6-well plate and 2 × 1010 library viral particles were applied. Total RNA was extracted from Mo-DCs 36 h later using RNeasy Kit. Earlier extraction time point was used to avoid biases due to differences in expression efficiency of library members (Qiagen, #74004) (Fig. 1A). Even though the variants are transcribed under the same constitutive promoter, such differences in expression could stem from codon optimality and GC content bias, as shown previously [29, 30].
Adult dermal fibroblasts were purchased from Millipore Sigma (106-05 A) and cultured in fibroblast growth medium (Millipore Sigma, 116-500) at 37 °C and 5% CO2. Once outgrown, 2 × 10^6 cells were infected with 2 × 10^10 library viral particles. Total RNA was extracted from dermal fibroblasts the next day using RNeasy Kit (Qiagen, #74004) (Fig. 1A).
Animal experimentsCanine (dog) experiments were performed at Absorption Systems California, LLC, (San Diego) labs (ASC protocol). The animal procedures were conducted in accordance with the guidelines of the National Institutes of Health (NIH) for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at Absorption Systems. The institution is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and maintains a current Public Health Service (PHS) Animal Welfare Assurance. A 1-year-old male Canine (Beagle) was injected with the AAV capsid library viruses described above; with 5E13vg/kg via intravenous route. Four weeks after injection, the Beagle was euthanized and various tissues including heart muscles, liver, and brain were harvested & flash-frozen for tissue-specific chimeric capsid enrichment and further analysis. Total RNA was extracted from the liver and muscle tissues using RNeasy Kit (Qiagen, #74004) and RNeasy Fibrous Tissue Kit (Qiagen, #74704) respectively.
Enriched AAV capsid recovery from liver and muscle tissues, DCs, and fibroblastscDNA synthesis was performed using SuperScript IV reverse transcriptase (Thermo Fisher, 18090050) and with a pool of custom-designed 10-mer sequence-specific primers (Supplementary Table 1). To increase the stability of the long cDNAs ( ~ 2230 bp), we proceeded with second-strand synthesis (Invitrogen, #A48570). Capsid variants expressed mRNA sequences were amplified from the second strand cDNA samples, with inversely oriented BasI overhang containing primers binding to upstream FwdUniversalCapsid-BsaI (5’-gggtctcgATGATTTAAATCAGGTATG -3’) and downstream RevCap_V8_BsaI primer (5’-CCATACCACATTTGTAGAGGTTT-3’) using the Platinum™ SuperFi™ PCR Master Mix (Invitrogen, #12358010). PCR amplified full capsid sequences were column purified (Qiagen, # 28104) and digested with BsaI and ligated into RJB p495 vector backbone, transformed into bacteria for plasmid amplification and later digested with HindIII (digests the backbone containing the capsids sequences with some extra sequence pads for flexibility & to improve sequence quality) and gel purified the enriched capsid pool for PacBio Sequencing.
PacBio Sequencing and data generationBoth the initial pooled chimeric virus samples and tissue-specific (Liver & Muscle) enriched capsid samples were used for PacBio long-read sequencing by Azenta, USA, and Histogenetics, USA respectively.
AAV sample preparation and sequencing were done by Yale Center for Genomic Analysis according to PacBio’s SMRTbell prep kit 3.0. Briefly, 7e11 vector genomes were treated with 20 U of DNase I (NEB M0303S) in a 200 μL vol for 10 min at 37 °C. DNA was extracted by using PureLinkTM Viral RNA/DNA Mini Kit (Thermo Fisher Scientific 12280050) following the manufacturer’s instructions. Samples have undergone a thermal annealing step by heating in annealing buffer (25 mM NaCl, 10 mM Tris-HCl [pH 8.5], 0.5 mM EDTA [pH 8]) at 95 °C for 5 min and then cooling to 25 °C (1 min for every −1 °C) on a thermocycler (Eppendorf Mastercycler). Libraries for vector DNA were constructed using the SMRTbell prep kit 3.0 by first performing end repair and A-tailing at 37 °C for 30 min followed by 65 °C for 5 min and subsequent addition of 4 μL of adapter per sample and Incubation at 20 °C for 30 min. Libraries were pooled and purified using 1.3X SMRTbell cleanup beads. Samples were then incubated with 5 ul of Nuclease for 15 min at 37 °C and purified again using 1.3X SMRTbell cleanup beads. Libraries were evaluated by Qubit dsDNA HS (Thermo Fisher Scientific Q32851) assay kit as well as using a DNA 7500 Bioanalyzer kit (Agilent 5067-1506). Sequencing was performed on a Sequel II instrument following standard procedures defined by the manufacturer and the Yale Center for Genomic Analysis.
Synthetic datasetsFor validation of the algorithm, we produced synthetic data of chimeric variants, simulating random fragmentation, homologous recombination, and PacBio sequencing events. First, we performed multiple sequence alignment of the 16 parental AAV serotypes, including AAV1-13, AAVrh8, AAVrh10, and AAVrh32, using the Clustal Omega program (v1.2.4) (with the options --outfmt clu --force) [28]. This information on the regions of homology was then used to simulate the DNA shuffling process. Consecutive fragments produced based on randomly chosen boundary positions in randomly chosen serotypes were concatenated to produce the chimeric variants. As PCR biases, partial digests, or varying homologous recombination and packaging efficiencies can favor certain serotypes, we assigned different probabilities to the serotype selection to mimic real-world settings as follows: AAV2 (25%), AAVrh8 (20%), AAV6 (17%), AAV7 (10%), AAV9 (7%), AAV8 (6%), AAV13 (6%), AAV1 (5%), AAV5 (2%), AAV3 (1%), AAVrh10 (1%), AAVrh32 (0%), AAV4 (0%), AAV10 (0%), AAV11 (0%), and AAV12 (0%). The fragment lengths were chosen from a uniform distribution defined within the 100-700 bp range, to reflect the usual size selection during the experimental workflows, as larger fragments can reduce diversity and shorter fragments are less prone to proper annealing [16, 31]. The information about parental compositions of the derived chimeric sequences was stored as ground truth for measuring the accuracy of our algorithm (Supplementary Fig. 1).
This method was used to generate 300 distinct chimeric sequences. Each sequence was assigned an abundance value to mimic the redundancy of sequences in the library of chimeric variants resembling the experimental settings. Specifically, 80% of chimeric sequences were randomly assigned an abundance value of 1. For the remaining 20% of chimeric sequences, abundance values were sampled from a normal distribution with a mean of 1 and a standard deviation of 50, while disregarding any values below 1.5. The SimLoRD program (v1.0.4) [32] was used to simulate PacBio circular consensus sequences (CCS) by taking each generated chimeric sequence as a reference (-rr) and its abundance count as the number of simulated reads (-n). The entire data simulation process was replicated five times to generate five different datasets using different seed values for the R functions involving random sampling.
Published AAV vector dataThe amino acid sequences of AAV vectors NP59 [33], 10A1-KP1 [34], Patent Number WO2019191701A1], and AAV-SYD12 [14] were obtained to explore parental AAV serotype contributions within these vectors.
Comments (0)