Patient blood specimens and cell lines
Six pairs of blood samples were obtained from patients with HCC before and after 125I seed implantation therapy for exosome extraction and RNA sequencing (RNA-seq). This study was approved by the Ethics Committee of the Sun Yat-sen University Cancer Center. Written informed consent was obtained from all participants of the study.
For the 125I seed implantation procedure, computed tomography (CT)-guided percutaneous puncture implantation was used. Briefly, preoperative localization was performed with a 16-multidetector-row CT scanner (Brilliance CT BigBore; Phillip Medical Systems, the Netherlands) guidance. After determining the entry point and path of the needles, 18-gauge needles were inserted into the tumors at intervals of at least 1.0 cm according to the preoperative plan. Precautions were taken to avoid the puncture of large blood vessels and important organs. After the needles were placed at the predetermined position, the 125I seeds were implanted at 0.5 cm intervals. Bleeding, pneumothorax and other complications were excluded by rescanning CT. The clinical information of patients undergoing RNA sequencing has been compiled in Additional file 1: Table S1.
PLC/PRF/5 and HepG2 HCC cell lines were purchased from Cellcook (Guangdong, China), cultured in minimum essential medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (ExCell Biological Technology, Suzhou, China), 1% penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA), and 1% non-essential amino acids (ELGBIO Biotechnology, Guangdong, China) and maintained in a humidified cell incubator at 37 ℃ and 5% CO2. All cells were tested using a MycoBlue Mycoplasma Detector (Vazyme Biotech, Nanjing, China) and confirmed to be mycoplasma-negative before use in the experiments.
Exosome extraction
The exosomes were extracted by ultracentrifugation. Briefly, plasma samples were collected from patients and centrifuged for 30 min at 500 × g. Supernatants were centrifuged at different speeds (2,000 × g, 10,000 × g, and 100,000 × g) to remove dead cells and cell fragments and obtain exosomes and interfering proteins. Subsequently, the pellet was resuspended in phosphate-buffered saline (PBS) and centrifuged at 100,000 × g for 70 min. This experiment was performed at 4 ℃. Finally, PBS was used to resuspend the pellet before storage at –80℃ for further experiments.
RNA-seq
The RNA-seq experiments were conducted by Epibiotech Co., Ltd. Briefly, RNA from plasma exosomes was extracted using the exoRNeasy Serum/Plasma Maxi kit (Qiagen, Hilden, Germany). Subsequently, the RNA was reverse-transcribed to cDNA using Evo M-MLV RT Master Mix (Accurate Biology, Changsha, China). The reverse transcription process involved incubation at 37 °C for 20 min, followed by denaturation at 85 °C for 30 s. RNA sequencing was conducted utilizing the Illumina HiSeq X10/Hiseq 4000 platform. The expression values of circRNAs were predicted based on the anticipated circRNA sequences. This prediction was achieved by tallying the reads associated with circRNAs, encompassing both TopHat mapping and TopHat-fusion mapping reads. The expression levels were then quantified as RPM (Reads Per Million mapped reads). A heat map was drawn based on the expression values obtained.
Quantitative real-time PCR (qRT-PCR)
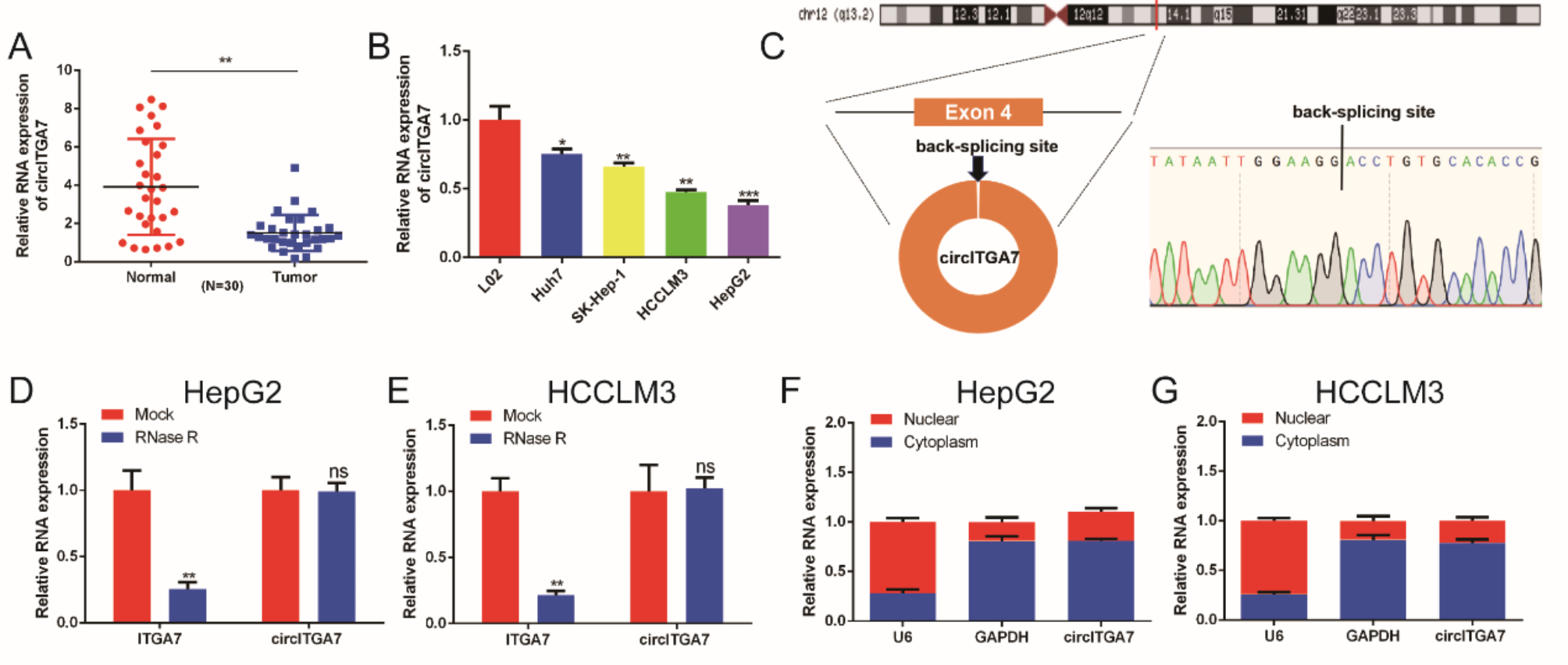
An RNA-Quick Purification Kit (Yishan Biotechnology, Shanghai, China) was used to extract total RNA from the cell lines. After measuring the RNA concentration using a NanoDrop 3000 spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), RNA was reverse-transcribed to cDNA using Evo M-MLV RT Master Mix (Accurate Biology). The reverse transcription was carried out at 37 °C for 20 min, followed by denaturation at 85 °C for 30 s. qRT-PCR analysis was performed using the LightCycler 480 System (Roche, Basel, Switzerland) using iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA). The specific temperatures and times for each step were as follows: initial denaturation at 95 °C for 5 min; amplification with 45 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 10 s, and extension at 72 °C for 10 s; melt curve analysis at 95 °C for 5 s, 65 °C for 1 min, and 97 °C continuous; and cooling at 40 °C. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and U6 was used as endogenous control to calculate relative mRNA or circRNA expression with the 2–ΔΔCT method. All PCR primer sequences are listed in Additional file 1: Table S2.
circRNA fluorescence in situ hybridization (FISH)
Cy3-labeled circEYA3 probes were obtained from Geneseed Biotech (Guangdong, China). FISH assay was performed using the HepG2 cell line. Briefly, cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. After drying, the pre-denatured (100 μM) probes were added to the culture well and incubated overnight at 37℃ in a humidified atmosphere. The slides were then rinsed thrice with saline sodium citrate buffer (Yaneng Bioscience, Shenzhen, China), followed by DAPI staining. Images were captured using the FV1000 confocal laser scanning microscope (Olympus, Tokyo, Japan). FISH probe sequence is as follows: 5' TTCACAATCAAAAGGAGGTAGTC 3'.
Nuclear-cytoplasmic fractionation
Nuclear and cytoplasmic RNA isolation was performed using Cytoplasmic & Nuclear RNA Purification Kit (AmyJet Scientific, Wuhan, China). Briefly, the cells were lysed with Lysis Buffer J and centrifuged at 14,000 × g for 10 min. The supernatant was transferred to a RNase-free centrifuge tube, and Buffer SK was added to both the supernatant and the pellet. The resulting mixture was then introduced into a centrifugal column and centrifuged at 3500 × g for 1 min. Following three washes of the centrifugal column with Wash Solution, RNA was eluted using Elution Buffer for subsequent qRT-PCR experiments.
RNase R treatment
Total RNA (5 μg) was extracted as mentioned above and treated with 3 U/μg RNase R (Geneseed Biotech, Guangzhou, China) and incubated for 30 min at room temperature. Then, the treated RNAs were incubated at 70 ℃ for 10 min to inactivate the enzyme. Relative RNA abundance was detected by qRT-PCR using circEYA3 and EYA3 linear mRNA primers.
Plasmid vector construction and transient transfection
The overexpression plasmids of circEYA3 were constructed by Geneseed Biotech. Overexpression plasmids for IGF2BP2 and short hairpin RNAs (shRNAs) against IGF2BP2 were obtained from Genechem (Shanghai, China). These sequences are listed in Additional file 1: Table S2. For transient transfection, Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific) was used according to the manufacturer’s instructions. Briefly, the transfection mixture, plasmids, and Opti-MEM I Reagent (Thermo Fisher Scientific) were added to 6-well plates when cell confluence reached 70–80%. The transfection medium was replaced 6–8 h later with normal medium.
Animal experiments
Male 4-week-old nude mice were purchased from Vital River (Beijing, China). All animals were treated in accordance with the guidelines of the Committee on Animals of Sun Yat-sen University. A subcutaneous xenograft tumor model was constructed as previously described. Briefly, 1 × 106 cells were subcutaneously injected into the right flank of the mice. When the average tumor diameter reached 5 ± 1 mm, 125I seeds were implanted. Under ultrasound guidance, a dedicated seed implantation needle was precisely positioned within the tumors. Subsequently, a 125I seed applicator was employed to facilitate the implantation of seeds (0.8 mCi per seed) at the desired locations of the tumors. Seven days after the implantation of 125I seeds, the circEYA3 overexpression plasmid (15 μg/mouse/injection) was injected into multiple sites within the tumor every 4 days for a total of three injections. Tumor tissues were collected on day 21.
Immunofluorescence (IF)
Briefly, cells were fixed and permeabilized as described above. The cells were washed twice with PBS and blocked with 5% bovine serum albumin. γH2A.X antibody (#9718, Abcam, Cambridge, MA, USA) and the corresponding secondary antibody were incubated with cells sequentially, followed by staining with DAPI (KeyGEN BioTECH, Nanjing, China). Finally, fluorescence images were captured using FV1000 confocal laser scanning microscope (Olympus).
Single cell gel electrophoresis (SCGE)
A CometAssay SCGE Kit (R&D Systems, Tustin, CA, USA) was used. Briefly, 2 h after irradiation or continuous 125I exposure for 2 days, the cells were resuspended in PBS and mixed with low-melting-point agarose gel. The mixture was then spread onto the solidified normal-melting point agarose gel and allowed to stay for 30 min at 4 ℃ in the dark. After immersion in the lysis buffer, the slides were subjected to electrophoresis at 25 V for 30 min. Next, 40 μL propidium iodide (PI) was added for staining and observed using the ECLIPS Ti-2 inverted microscope (Nikon, Tokyo, Japan).
Cell proliferation assays
Cell viability was measured every 24 h for 5 consecutive days using the Cell Counting Kit-8 (CCK-8, Yishan Biotechmology, Shanghai, China). Transfected cells were seeded into 96-well plates at a density of 5 × 103/well with or without irradiation exposure. After incubation with the CCK-8 assay solution for 2 h, the absorbance of the cells in the microplate reader (BioTek Instruments, Winooski, VT, USA) was determined at 450 nm. Cell viability was normalized to the absorbance on day 1.
Flow cytometry
To measure the apoptosis rate, the Annexin V-AF647/PI Apoptosis Kit (Yishan Biotechnology) was used. Cells seeded in 6-well plates were digested with 0.25% trypsin, centrifuged, and resuspended in binding buffer. Then, 10 μL Annexin V-AF647 and 5 μL PI were added to the cell suspension followed by incubation for 5 min in the dark. Flow cytometry was then performed using the CytoFLEX flow cytometer (Beckman Coulter, Pasadena, CA, USA).
RNA pulldown assay
RNA pulldown assay was performed to identify circEYA3-binding proteins, according to the manufacturer’s protocol. Briefly, by incubating the cell lysates with streptavidin-coated magnetic beads, the biotin-coupled RNA complex was pulled down, and the combined protein was eluted from the filled beads. Subsequently, western blotting (WB) and mass spectrometry analyses were performed.
RNA immunoprecipitation (RIP)
RIP assay was conducted using the IEMed-K303 RIP Kit (IEMed, Guangzhou, China) according to the manufacturer’s instructions. Briefly, the cells were lysed with lysis buffer and incubated with an anti-IGF2BP2 antibody or negative control immunoglobulin G overnight. Magnetic beads were then added, and the cells were resuspended with Proteinase K to remove proteins. RNA was purified and analyzed using qRT-PCR.
WB
Proteins were extracted from the cell lines using radioimmunoprecipitation lysis buffer containing phosphatase and protease inhibitors. After the protein concentration was measured using the BCA Protein Quantitation Assay Kit (KeyGEN), equal amounts of total proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (ACE Biotechnology, Nanjing, China) and transferred to polyvinylidene difluoride membranes (Millipore, Burlington, MA, USA). The membranes were incubated with the corresponding primary and secondary antibodies, followed by detection with enhanced chemiluminescence (GBCBIO, Guangdong China) using the ChemiDoc MP Imaging System (Bio-Rad Laboratories), with GAPDH as a control. The primary antibodies used were anti-GAPDH (1:5000; Proteintech, Shanghai, China), anti-IGF2BP2 (1:5000; Proteintech), anti-DTX3L (1:600; Proteintech), anti-γH2A.X (1:1000, Cell Signaling Technology, MA, USA), anti-caspase-3 (1:1000, Cell Signaling Technology), and anti-cleaved caspase-3 (1:1000, Cell Signaling Technology).
Statistical analysis
GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA), SPSS version 23.0 (IBM, Armonk, NY, USA), and R version 4.1.0 (R Foundation for Statistical Computing, Vienna, Austria) were used for statistical analysis. If the data adheres to a normal distribution, it is expressed as mean ± standard deviation and subjected to analysis using the Student’s t-test. In cases where normal distribution is not met, representation is done as median with interquartile range, and analysis is performed using the Wilcoxon test. All tests were conducted as two-sided tests and p ≤ 0.05 was considered statistically significant. All data were generated by at least triplicate independent experiments.





Comments (0)