
Remember me

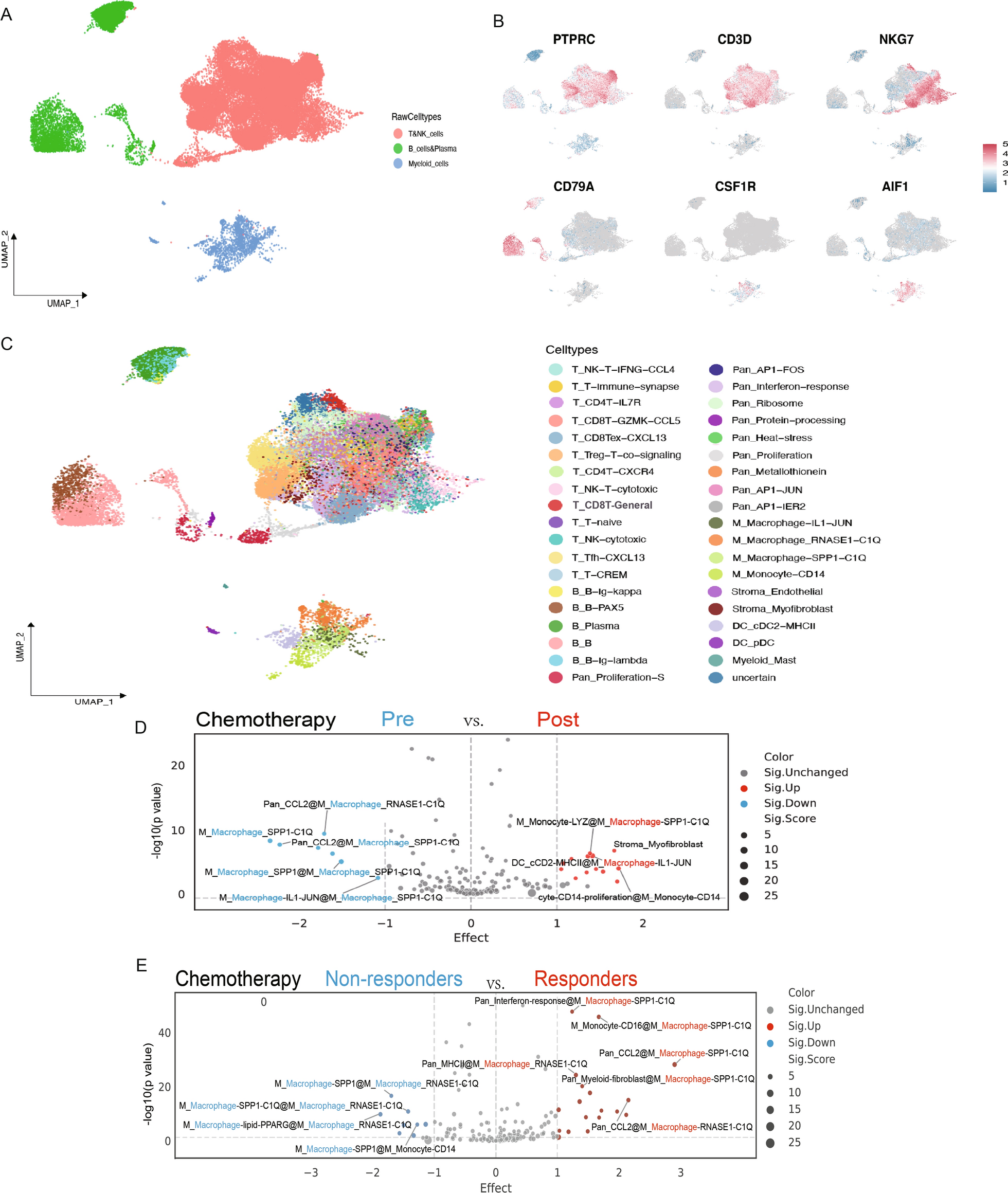



To identify tumor infiltrating lymphocyte (TIL) subsets associated with anti-tumor immunity in BC, we reanalyzed scRNA-seq datasets from BC samples [11] using the recently developed data-driven framework MetaTiME [15]. This method addresses limitations in resolution and consistency inherent in manual annotation by known gene markers, allowing for a comprehensive and continuous characterization of cell states.
Initially, we categorized the single cells into three primary immune cell clusters based on classical marker genes: myeloid cells (marked by CSF1R and AIF1), T cells and natural killer (NK) cells (marked by CD3D, PTPRC, and NKG7), and B cells and plasma cells (marked by CD79A) (Fig. 1A and B; Supplementary Fig. 1).
Fig. 1
TAM as a Major Contributor to BC Clinical Outcomes. A UMAP visualization depicts three primary immune cell clusters from BC (n = 5) tissues, including T cells & NK cells, B cells & plasma cells, and myeloid cells. B Expression of classical marker genes used to identify the main clusters (myeloid cells: CSF1R, AIF1; T & NK cells: CD3D, PTPRC, NKG7; B & plasma cells: CD79A). C MetaTiME cell state annotations based on top-enriched MeC in BC cell clusters. D Differential enrichment of cell states in BC pre- and post-treatment, evaluated using MeC feature testing. X-axis: difference in average feature scores between pre- and post-chemotherapy conditions. Y-axis: -log(p-value) from two-sided t-tests. Significant cluster difference signatures are labeled as “EnrichedMeC@ClusterName”; when the enriched MeC coincides with the cluster name, the signature is marked as “ClusterName”. Red dots represent signatures with increased difference; dot size is proportional to the average feature score of cells in the cluster under post-immunotherapy conditions. Blue dots indicate suppressed features; dot size is proportional to the average feature score of cells in the cluster under pre-chemotherapy conditions. E Differential enrichment of cell states between BC treatment responders and non-responders, assessed using MeC feature testing
Using MetaTiME, we further annotated these three primary immune cell clusters, subdividing them into 37 distinct immune cell subclasses (Fig. 1C; Supplementary Table 2). Given the variability in cell type proportions and the small number of patients in each cohort, we focused on changes in MeC characteristics rather than absolute cell count proportions to better understand immune responses following tumor treatment.
We analyzed samples from the same BC cohort collected both before and after treatment (Fig. 1D; Supplementary Table 3) and compared treatment responders to non-responders (Fig. 1E; Supplementary Table 4). Notably, TAMs exhibited the most pronounced dynamic changes in MeC characteristics, irrespective of treatment duration or response status.
These dynamic changes were primarily concentrated in a specific TAM subgroup characterized by high expression of SPP1 and C1QA, which we designated as M_Macrophage-SPP1-C1Q. Quantitative comparisons reinforced this observation, showing consistent changes in this subgroup across different conditions (Supplementary Figs. 2A and 2B).
In summary, our findings highlight the significant functional complexity and dynamic nature of TAMs in the BC TME. The pronounced changes observed in the M_Macrophage-SPP1-C1Q subgroup suggest that it may play a crucial role in influencing prognosis and treatment efficacy in BC patients.
Functional characteristics of TAM subgroups in BCTo investigate the functional heterogeneity of TAM subgroups in BC, we analyzed DEGs within each cluster (Supplementary Fig. 2E, Supplementary Table 5). Enrichment analysis of classical M1 and M2 macrophage gene signatures revealed distinct patterns among the three TAM clusters. The M_Macrophage-IL1-JUN cluster exhibited high enrichment for M1-associated genes, indicative of a pro-inflammatory phenotype, while the M_Macrophage-RNASE1-C1Q cluster showed higher enrichment for M2-associated genes, reflecting an anti-inflammatory or tissue-repair phenotype. By contrast, the M_Macrophage-SPP1-C1Q cluster displayed low enrichment for both M1 and M2 signatures, suggesting it does not conform to the traditional M1/M2 classification (Fig. 2A). Further GSEA demonstrated that M_Macrophage-SPP1-C1Q was enriched in cell proliferation pathways such as MTORC1 signaling and MYC targets, as well as oncogenic pathways including KRAS signaling, P53 pathway, and epithelial-mesenchymal transition (EMT) (Fig. 2B). These findings highlight the distinct proliferative and oncogenic properties of this TAM subgroup.
Fig. 2
Functional Characteristics of TAM Subgroups in BC. A Scatter plot displaying the enrichment scores for M1- and M2-macrophage gene signatures across three TAM subsets. B GSEA heatmap illustrating the enrichment of specific gene sets in each TAM subgroup. Blue cells indicate no statistically significant enrichment, while red cells indicate statistically significant enrichment (P < 0.05). The number of asterisks within a cell denotes the level of statistical significance, with more asterisks corresponding to smaller P-values. The dendrogram on the left clusters gene sets based on their expression patterns across TAM subgroups, while the bar at the top indicates whether the enriched gene sets are upregulated or downregulated in each subgroup. C Density heatmap illustrating tumor-related functional activities enriched in different TAM subgroups. The color gradient represents the density of enrichment, with red indicating higher density and blue indicating lower density. The dashed lines represent statistical distribution summaries: the mean (solid black line), the 25th percentile, the 50th percentile (median), and the 75th percentile of the data for each subgroup. D Box plots showing stemness scores of different TAM subgroups. E Potential polarization trajectories of different macrophage subgroups inferred by Monocle 2. F Visualization of functional characteristics and gene changes in TAM subgroups among responders and non-responders using the ClusterGVis package
To delve deeper into the unique features of M_Macrophage-SPP1-C1Q, we examined the enrichment of specific functional pathways within this cluster. It showed high enrichment in cell proliferation (MTORC1 signaling, MYC targets V1), inflammatory responses, complement signaling pathways, oxidative phosphorylation, and interferon response (Fig. 2C). These results further support the notion that M_Macrophage-SPP1-C1Q has a unique functional profile distinct from traditional TAM subsets.
We next explored the unique features of M_Macrophage-SPP1-C1Q by analyzing its functional pathway enrichment. This cluster showed high enrichment in cell proliferation (MTORC1 signaling, MYC targets V1), inflammatory responses, complement signaling, oxidative phosphorylation, and interferon response pathways (Fig. 2C), reinforcing its distinct functional profile. Further, stemness analysis using cytoTRACE revealed that M_Macrophage-SPP1-C1Q exhibited the highest developmental potential among the three TAM clusters, while M_Macrophage-RNASE1-C1Q exhibited the lowest (Fig. 2D; Supplementary Fig. 2F). Differentiation trajectory analysis using Monocle suggested that M_Macrophage-SPP1-C1Q serves as an intermediate transitional state in TAM polarization, while M_Macrophage-RNASE1-C1Q and M_Macrophage-IL1-JUN occupied opposite ends of the trajectory, representing more differentiated states (Fig. 2E). This result suggests that M_Macrophage-SPP1-C1Q may serve as a key transitional state in TAM dynamics.
In the context of treatment response, M_Macrophage-SPP1-C1Q was enriched in genes and pathways associated with oncogenesis and an inhibitory TME, such as TGFB1, SPP1 [20], FABP5 [21], SLC11A1 [22], and VCAN [23], in treatment responders (Supplementary Fig. 2E). Meanwhile, M_Macrophage-RNASE1-C1Q was linked to immunoglobulin-mediated immune responses and B cell-mediated immunity, promoting an anti-tumor TME [24, 25], while M_Macrophage-IL1-JUN was associated with apoptosis-related functions. In non-responders, M_Macrophage-SPP1-C1Q was enriched in pathways related to neutrophil chemotaxis and migration, which have been implicated in chemotherapy resistance through TGF-β activation [26]. Conversely, M_Macrophage-RNASE1-C1Q was enriched in complement activation pathways [27], known for promoting immunosuppression and tumor progression, while M_Macrophage-IL1-JUN was enriched in cell development and growth-related pathways (Fig. 2F).
These results demonstrate that the functional characteristics and differentiation potential of TAM subgroups, particularly the unique properties of M_Macrophage-SPP1-C1Q, significantly influence the TME and treatment outcomes in BC.
High infiltration of M_macrophage-SPP1-C1Q is associated with poor prognosis in BCTAMs play a pivotal role in tumor progression and response to therapy. To investigate the prognostic significance of TAM subgroups in BC, we employed CellChat analysis to examine potential ligand-receptor interactions among the main TIIC subgroups in treatment responders and non-responders. In the responder group, M_Macrophage-RNASE1-C1Q and M_Macrophage-SPP1-C1Q demonstrated strong interactions with other immune cells, particularly as predominant ligand-expressing cell types (Fig. 3A). This suggests that these TAM subgroups may actively communicate with immune cells in the TME, influencing anti-tumor immune responses [28]. Interestingly, M_Macrophage-SPP1-C1Q showed a higher signal-sending propensity in both responders and non-responders, with a more pronounced effect in non-responders (Fig. 3B), indicating its potential role in modulating the TME, particularly under conditions of treatment resistance.
Fig. 3
High Infiltration of M_Macrophage-SPP1-C1Q in BC Correlates with Poor Prognosis. A Heatmap displaying the relative expression levels of ligand-receptor pairs among all immune cells in treatment responders compared to non-responders. B Key ligand-receptor pairs between different treatment response groups. Dot size represents the probability of communication for specific ligand-receptor pairs between sender and receiver clusters. C Heatmap showing Spearman correlation coefficients between proportions of different cell subsets. D Forest plot illustrating univariate Cox regression analysis of OS for each TAM subset in the GSE58812 cohort. E Infiltration abundance of M_Macrophage-SPP1-C1Q in chemotherapy-sensitive and chemotherapy-resistant patients. F Infiltration abundance of M_Macrophage-SPP1-C1Q in patients with and without metastasis. G, H Kaplan–Meier survival curves for MFS (left) and OS (right) among patients with high and low M_Macrophage-SPP1-C1Q infiltration in the GSE58812 and METABRIC cohorts
Further exploration of TAM interactions revealed that M_Macrophage-IL1-JUN exhibited strong correlations with regulatory T cells (Tregs) and naive T cells, suggesting its involvement in creating an immunosuppressive TME (Fig. 3C). Conversely, M_Macrophage-SPP1-C1Q displayed significant correlations with broader functional gene sets, including Pan-Metallothionein and Pan-Proliferation, as well as with dendritic cell subsets DC_pDC and DC_cDC2-MHCII, reflecting its distinct immunological role in the TME. These findings highlight the heterogeneity and complex immunological functions of TAM subgroups in BC.
To assess the clinical relevance of M_Macrophage-SPP1-C1Q, we evaluated its infiltration levels in a cohort of 107 BC patients using ssGSEA and scRNA-seq-based overexpression signatures (Fig. 3D; Supplementary Table 7). Notably, M_Macrophage-SPP1-C1Q infiltration was significantly higher in chemotherapy-resistant patients compared to chemotherapy-sensitive patients (Fig. 3E). Similarly, patients with metastatic tumors exhibited higher infiltration levels of M_Macrophage-SPP1-C1Q than those with primary tumors (Fig. 3F), suggesting a strong association between M_Macrophage-SPP1-C1Q and treatment resistance. Furthermore, univariate Cox regression analysis revealed that M_Macrophage-SPP1-C1Q infiltration was significantly associated with overall survival (OS), while survival analyses demonstrated that patients with higher infiltration levels of this subgroup had significantly reduced OS and shorter metastasis-free survival (MFS) (Fig. 3G). These findings were validated in the METABRIC dataset, which confirmed the association between high M_Macrophage-SPP1-C1Q infiltration and poor survival outcomes in an independent BC cohort.
These results suggest that high infiltration of M_Macrophage-SPP1-C1Q is strongly associated with treatment resistance, reduced survival, and poorer clinical outcomes in BC.
M_macrophage-SPP1-C1Q identifies clinically distinct subgroups of BC patientsThe inherent heterogeneity of BC necessitates effective stratification of patients into clinically meaningful subgroups. To explore the potential of M_Macrophage-SPP1-C1Q as a marker for such stratification, we identified 37 genes uniquely associated with this TAM subtype, referred to as M_Macrophage-SPP1-C1Q-specific core metagenes (Supplementary Tables 5 and 6, Fig. 4A). These genes, representing the distinctive transcriptional signature of the subtype, were used for consensus clustering analysis in the METABRIC BC cohort. This analysis classified patients into two distinct molecular subtypes, termed LM (Low Metagene expression) and HM (High Metagene expression) (Fig. 4B). The robustness of these clusters was validated using t-distributed stochastic neighbor embedding (t-SNE) and spectral pattern clustering, which confirmed consistent subgrouping (Supplementary Fig. 3A).
Fig. 4
Identification of Potential Macrophage Subtypes in BC. A Venn diagram of core metagenes for M_Macrophage-SPP1-C1Q. B Consensus clustering in the METABRIC cohort based on 37 core metagenes. C Box plot illustrating the varying estimated proportions of M_Macrophage-SPP1-C1Q across different subtypes. D Heatmap of core metagenes of M_Macrophage-SPP1-C1Q and clinical features. E Alluvial diagram depicting the interrelations among BC patient subtypes, survival status, and Claudin subtype. F, G Kaplan–Meier analysis of OS (F) and relapse-free period (G) for BC patients belonging to two different molecular clusters. H Univariate and multivariate analyses of the association between clinical-pathological features and macrophage subtypes in the METABRIC cohort
Patients in the HM cluster exhibited significantly higher expression of the 37 core metagenes compared to those in the LM cluster (Fig. 4D). Correspondingly, ssGSEA analysis demonstrated a higher infiltration abundance of M_Macrophage-SPP1-C1Q in the HM cluster (Fig. 4C), indicating that this subgroup is enriched with the M_Macrophage-SPP1-C1Q subtype within the TME. Survival analysis revealed that patients in the HM cluster had significantly poorer outcomes, with reduced OS and shorter relapse-free periods compared to patients in the LM cluster (Fig. 4F and G; Supplementary Fig. 3B). This suggests that high expression of M_Macrophage-SPP1-C1Q-specific core metagenes is closely associated with adverse clinical outcomes.
To further evaluate the clinical relevance of this classification, we examined its relationship with the Claudin Subtype, a recognized molecular classification in BC. Both LM and HM clusters included patients from the Claudin Subtype, with significant survival differences observed within these subgroups (Fig. 4E). Additionally, univariate and multivariate Cox regression analyses demonstrated that the HM cluster classification remained significantly associated with OS after adjusting for confounding factors such as age, tumor size, grade, and hormone receptor status (Fig. 4H). These findings underscore the prognostic value of the M_Macrophage-SPP1-C1Q-based subtype classification as an independent predictor of patient outcomes.
These results demonstrate that molecular subtyping of BC patients based on M_Macrophage-SPP1-C1Q-specific core metagenes effectively stratifies patients into clinically distinct subgroups. The identification of the HM cluster, characterized by high infiltration of M_Macrophage-SPP1-C1Q and associated with poorer prognoses.
Development of an M_macrophage-SPP1-C1Q derived subtype system for predicting patient survival benefitTo evaluate the clinical utility of the M_Macrophage-SPP1-C1Q molecular subtype in predicting patient outcomes, we compiled bulk RNA-seq data and clinical information from multiple BC cohorts. These datasets were divided into a training set (n = 529), a test set (n = 228), an internal validation set (n = 325), and an external validation set (n = 1,980). Using the expression profiles of the 37 M_Macrophage-SPP1-C1Q-specific core metagenes, we developed predictive models based on seven machine learning algorithms, including NB, RF, KNN, LogiBoost, XGBoost, Elastic Net, and Lasso Regression (Fig. 5A and B). After a 10-repeated fivefold cross-validation to prevent overfitting, the RF model demonstrated the highest predictive accuracy with an AUC of 0.89 (Fig. 5C). This model, named the Macro.RF model, was selected as the final predictive tool.
Fig. 5
Development of the Macro.RF Derived Subtype System for Prognosis and Treatment Efficacy Prediction in BC. A Flowchart illustrating the construction of the Macro.RF model using machine learning processes. B Comparison of multiple ROC curves representing the performance of different machine learning algorithms in the validation set. C Schematic representation of molecular subtypes based on M_Macrophage-SPP1-C1Q. D ROC curve depicting the performance of the final Macro.RF model in validation and test cohorts. E Kaplan–Meier curves comparing OS between patients in the LM (good prognosis) and HM (poor prognosis) groups in the validation and test sets. F, G Clinical response rates to chemotherapy in patients stratified into LM (good response) and HM (poor response) groups across different cohorts
To further validate the Macro.RF model’s robustness and generalizability, we tested it on an independent dataset. The model consistently exhibited high predictive performance, with an AUC comparable to the training cohort (Fig. 5D). Survival analyses revealed that patients classified as belonging to the M_Macrophage-SPP1-C1Q subtype had significantly shorter OS compared to those in the non-M_Macrophage-SPP1-C1Q group (all p < 0.001, Fig. 5E), underscoring its ability to stratify patients based on survival risk. These findings highlight the model’s potential to predict clinical outcomes accurately.
We also investigated the utility of the Macro.RF model in predicting chemotherapy responses, given the critical role of chemotherapy in BC treatment. Applying the model to two independent chemotherapy cohorts (GSE14094 and GSE18728), we categorized patients into ‘good’ and ‘poor’ prognostic groups. In the GSE14094 cohort, the remission rate in the ‘good’ group was 40.1%, significantly higher than the 33% observed in the ‘poor’ group (Fig. 5F). Similarly, in the GSE18728 cohort, remission rates were 35.2% in the ‘good’ group versus 20.6% in the ‘poor’ group (Fig. 5G). These results demonstrate that the Macro.RF model effectively predicts chemotherapy response, providing valuable clinical insights for patient management.
In summary, the analysis confirmed that the Macro.RF model, based on the M_Macrophage-SPP1-C1Q core metagenes, effectively stratifies BC patients, reflecting its robust predictive capability in clinical settings.
Comments (0)