Tissue specimen collection
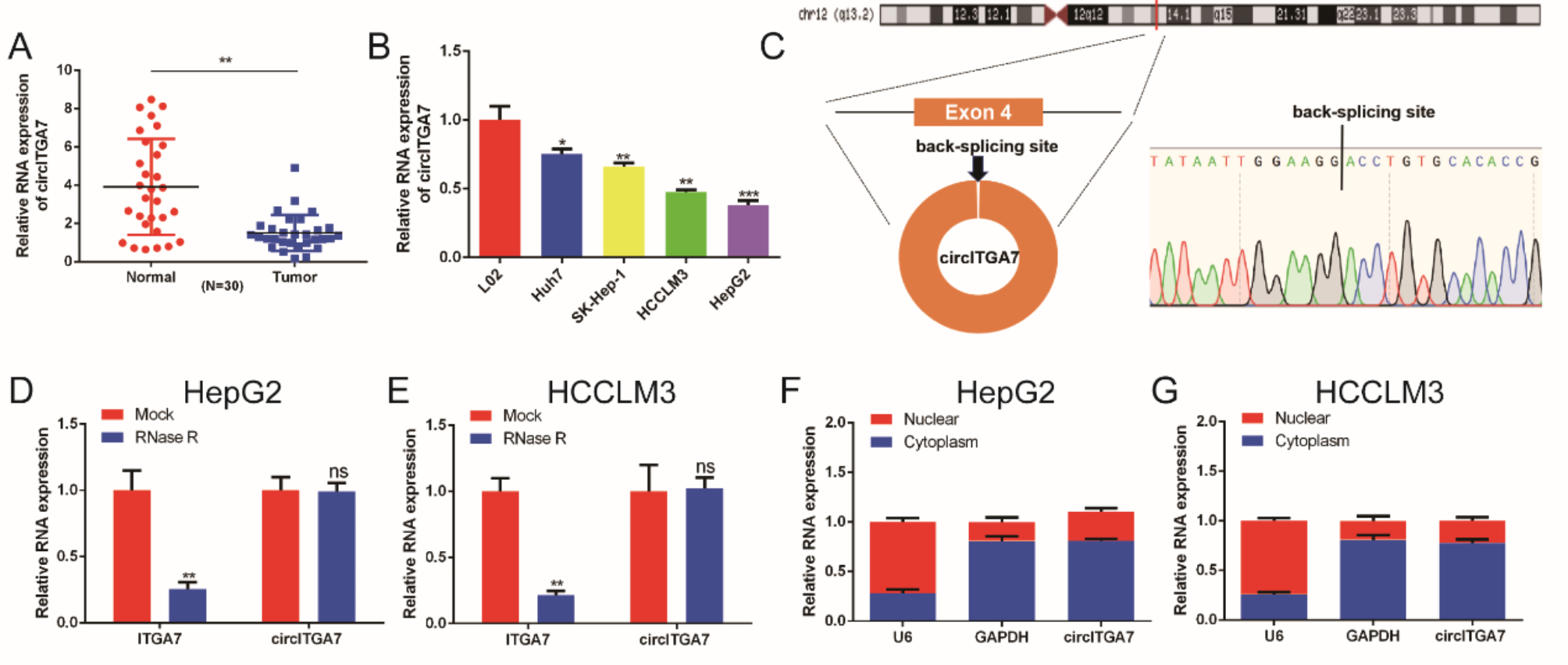
Fresh HCC tissues and adjacent non-tumor sites were obtained from 30 paired patients who were diagnosed with primary HCC before receiving any treatment between 2021 and 2024 at the 302 Hospital of the Chinese People’s Liberation Army. The histological and pathological reports of the specimens were diagnosed by at least two professional clinical pathologists. Immediately after surgical resection, all tissue specimens were rapidly frozen in liquid nitrogen and then stored at -80 °C. All specimens were obtained with the consent of the patients and this study was approved by the Ethics Committee of the 302 Hospital of the Chinese People’s Liberation Army.
Cell culture and transfection
HCC cell lines (HepG2, HCCLM3, Huh7, SK-Hep-1) and normal liver epithelial cells (L02) were purchased from BNCC (Beijing, China). HepG2, HCCLM3, Huh7 and SK-Hep-1 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin. L02 cells were cultured in RPMI-1640 (Gibco) medium supplemented with 20% fetal bovine serum and 1% penicillin/streptomycin. All cells were maintained in a humidified incubator at 37 °C with 5% CO2. Cell transfection was performed using Lipofectamine 3000 reagent (Invitrogen, USA) according to the manufacturer’s instructions.
RNA extraction and qRT-PCR
Total RNA was isolated from cells or tissues using TRIzol reagent (Thermo Fisher, USA) and cDNA was synthesized with EasyScript® First-Strand cDNA Synthesis SuperMix (Transgen, Beijing, China). Real-time PCR analysis was conducted using SuperReal Colorful Fluorescent Quantitative Pre-Mix Reagent (Tiangen, Beijing, China) on a Roche LightCycler 480 real-time PCR system (Roche, Basel, Switzerland), with gene expression normalized to GAPDH. Primers were listed in Supplementary Table S1.
RNase R treatment
1 µg RNA samples was incubated at 37℃ for 15 min with or without 5 U RNase R (Epicentre Technologies, Madison, WI, USA). Then, the RNA samples were analyzed by qRT-PCR.
Nuclear and cytoplasmic extraction
The PARIS™ Kit (AM1556, Thermo Fisher Scientific, Waltham, MA, USA) was utilized to separate the cytoplasmic and nuclear fractions of the cells. The cells were lysed in a cell fraction buffer while on ice for a 10-minute period, followed by a centrifugation step at 400× g for 5 min at 4 °C. The resulting supernatant was designated as the cytoplasmic fraction, while the nuclear pellets were obtained by rinsing with the cell fraction buffer.
Construction of plasmids, siRNAs and miRNA mimics
The overexpression plasmid construction of circITGA7(pcDNA-circITGA7) involves cloning the cDNA sequence of circITGA7 along with 500 bp sequences from both sides of its genomic location into the pcDNA3.1 vector. An empty pcDNA3.1 vector serves as the negative control (vector). For the dual-luciferase reporter assay, plasmids (pmirGLO-circITGA7-WT or pmirGLO-circITGA7-Mut, pmirGLO-BCL11B-WT or pmirGLO-BCL11B-Mut) are constructed by cloning the cDNA sequences of circITGA7 and BCL11B, as well as the mutant sequences of their cDNAs into the pmirGLO vector. All these plasmid vectors are synthesized by Generalbiol (Anhui, China). The siRNAs (si-circITGA7 and si-BCL11B) for knocking down circITGA7 (AATTGGAAGGACCTGTGCACA) and BCL11B (TGCTGACTAACGACGTCAAAATC), along with the corresponding negative control siRNA sequences and the mimic of miR-330 are synthesized by Generalbiol (Anhui, China).
Cell counting Kit-8 (CCK-8) and colony formation assay
For CCK-8 assay, HepG2 and HCCLM3 cells transfected with vector (control group) and pcDNA-circITGA7 were cultured in 96-well plates (1 × 104 cells per well). After transfection for 24, 48 and 72 h, each well was added with 10 µL CCK-8 reagent (Beyotime, Shanghai, China) and incubated for 2 h. The absorbance of the cells at 450 nm was measured with an enzymoleter (Themofisher, USA). The absorbance values are directly proportional to cell viability.
For colony formation assay, HepG2 and HCCLM3 cells transfected with different nucleic acid sequences were cultured in 6-well plates (1 × 103 cells per well) for 1–2 weeks. Then, colonies were washed, fixed, stained and photographed. Colonies with > 50 cells were counted manually using ImageJ. The number of colonies per well was averaged across replicates.
5-ethynyl-2’-deoxyuridine (EdU) assay
HepG2 and HCCLM3 cells transfected with different nucleic acid sequences were cultured in 12-well plates (15 × 104 cells per well). 48 h after transfection, followed the instructions of BeyoClick™ EdU-555 cell proliferation detection kit (Beyotime, Shanghai, China). Images were then taken using a confocal laser microscope (Olympus, Tokyo, Japan). The percentage of EdU-positive cells (proliferating cells) relative to the total nuclei (Hoechst staining) was calculated using ImageJ software.
Wound healing assay
3 × 104 HepG2 and HCCLM3 cells were seeded into a 24-well plate and after attachment, they were transfected with different nucleic acid sequences. The cells were serum-starved for 24 h, after which a scratch was made across the cell monolayer using a 200 µL pipette tip to create wounds of consistent length. The cells were then washed with PBS to remove loose cell debris. Each wound was imaged at 0 and 48 h using an inverted microscope (Olympus, Tokyo, Japan). Migration was quantified by measuring the reduction in wound area at 0 h and 48 h using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Cell cycle and cell apoptosis assay
For cell cycle analysis, HepG2 and HCCLM3 cells transfected with various test substances were cultured in a 12-well plate. After 48 h of transfection, propidium iodide (PI) staining was performed according to the instructions of the Cell Cycle and Apoptosis Assay Kit (Beyotime, Shanghai, China). Cells were collected using a flow cytometer (Beckman, USA) and cell cycle analysis was conducted using ModFit software (Verity Software House, USA).
For cell apoptosis detection, HepG2 and HCCLM3 cells transfected with different test substances were cultured in a 12-well plate. After 48 h of transfection, PI and FITC staining were performed according to the instructions of the Annexin V-FITC/PI Apoptosis Detection Kit (keygentec, Jiangsu, China). Cell collection and apoptosis analysis were conducted using a flow cytometer (Beckman, USA).
Dual-Luciferase reporter gene experiment
HepG2 and HCCLM3 cells were co-transfected with pmirGLO-circITGA7-WT, pmirGLO-circITGA7-Mut, pmirGLO-BCL11B-WT or pmirGLO-BCL11B-Mut and either miR-330 mimics or miR-NC for 48 h. The firefly and renilla luciferase activities in different groups of cells were measured using the Dual-Luciferase Reporter Assay Kit (Promega, Fitchburg, WI, USA).
Tumor xenograft formation
Male NCG mice aged 5–6 weeks were purchased from Hfkbio (Beijing, China). The experimental protocol was approved and the experimental animals were handled according to the ethical standards of Beijing Tiantan Hospital. The mice were randomly divided into two groups and injected subcutaneously on the right side of the back with cells transfected with either vector or pcDNA-circITGA7 plasmids. A total of 5 × 106 cells were injected to establish a subcutaneous xenograft model. The mice were monitored for 28 days, with tumor measurements taken twice weekly. On day 28, the tumors were excised and weighed. Immunohistochemical (IHC) experiments of the tumors were conducted by Servicebio (Wuhan, Wuhan).
Statistics analysis
Statistical analysis was performed using the mean ± standard deviation (SD) from three independent replicates. Student’s t-test was conducted using GraphPad Prism 7 software (GraphPad, San Diego, CA, USA). A p-value of < 0.05 was considered statistically significant, while p ≥ 0.05 was considered not statistically significant and marked as ns.





Comments (0)