
Remember me




The study workflow is constructed upon the data acquired from TCGA-COAD-READ and is divided into eight sequential steps, as illustrated in Fig. 1. Within the datasets, around 5600 DEGs and DE-lncRNAs were identified, with the number of down-regulated (DR) entities showing a relatively higher count compared to up-regulated (UR) DEGs and DE-lncRNAs (Additional file 1: Table S1).
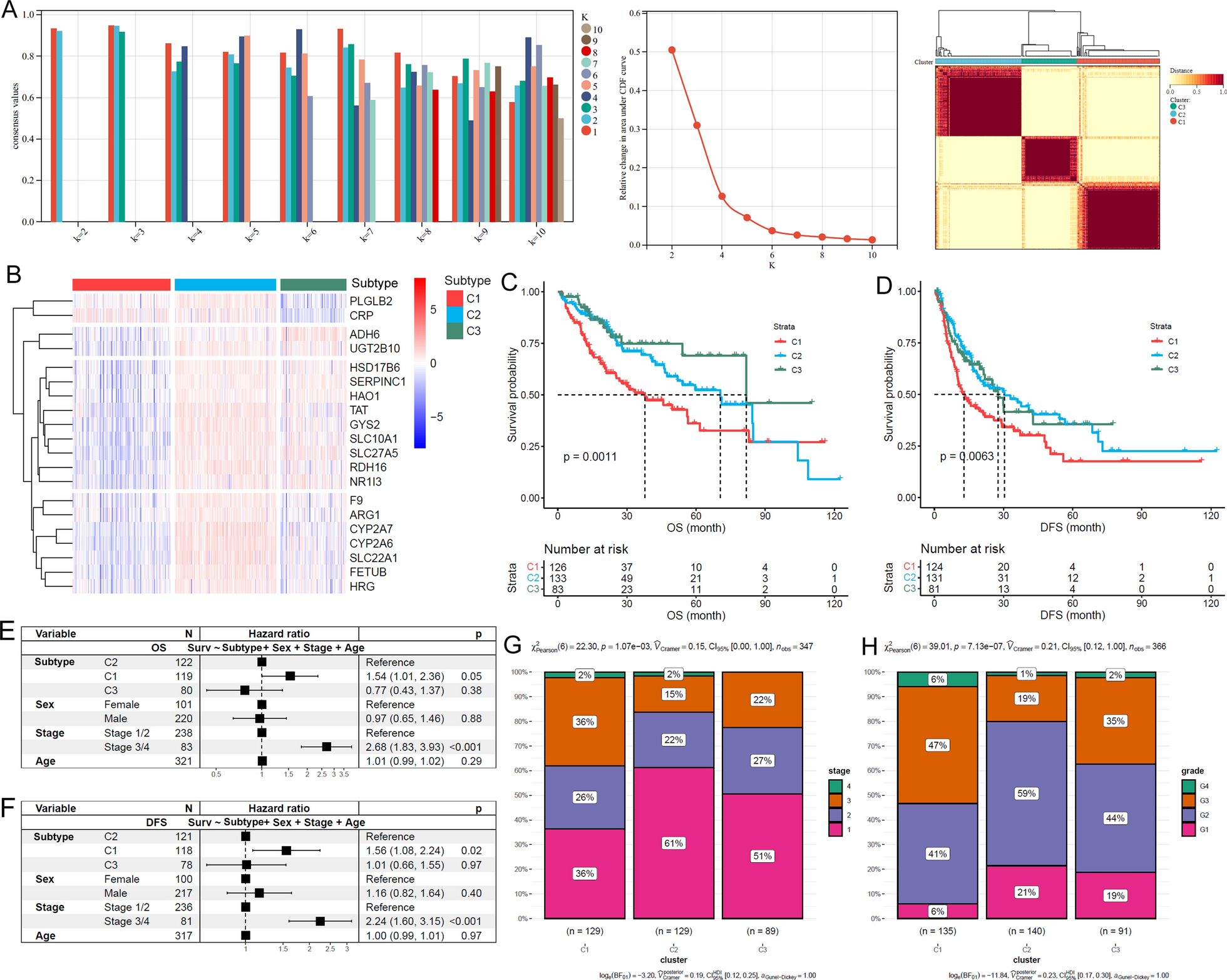
To construct a co-expression network using the TCGA COAD-READ dataset, a stepwise approach was employed. The initial step involved the creation of a sample clustering tree to identify and exclude outlier samples lacking of biological relevance. Subsequently, a soft-thresholding power of β = 3 (resulting in a scale-free R2 = 0.9) was set, resulting in the identification of fourteen distinct modules characterized by varying colors with non-clustering DEGs and DE-lncRNAs were designated as gray module (Fig. 2A–E). Furthermore, module-trait relationships were assessed (Fig. 2F), revealing that the blue module demonstrated heightened significance concerning clinical attributes associated with TNM staging (Cor = 0.47/p = 1.1e−154) (Fig. 2G). Within this context, a total of 2299 interacting DEGs and DE-lncRNAs within the blue module were selected for further in-depth analysis (Additional file 1: Table S2).
Fig. 2
The identification of pivotal modules demonstrating substantial associations with TNM staging within the TCGA COAD-READ dataset is outlined through the subsequent steps: A evaluation of the scale-free fit index (displayed on the left) and mean connectivity (presented on the right) across varying soft-threshold powers. B Presentation of an eigengene adjacency heatmap. C Depiction of a network heatmap plot featuring the principal modules. D Clustering of module eigengenes, with a distinctive red line marking the cut height (set at 0.3). E Formation of clustering dendrograms, aligning robust DEGs and DE-lncRNAs with their corresponding modules based on a unique dissimilarity measure (1-TOM). F Generation of a heatmap illustrating the correlation between module eigengenes and clinical attributes, specifically TNM staging, within the context of colorectal cancer (CRC). Each cell contains both the p-value and the correlation coefficient. G Illustration of a scatter plot emphasizing the paramount significance of the key module (blue module) in relation to CRC’s TNM staging
Identification of pivotal targets through analysis of the lncRNA–mRNA interacting networkTo construct a lncRNA–mRNA network, a total of highly correlated 604 DEGs and 22 DE-lncRNAs with potential interactions were used (Additional file 1: Table S3). Complex reciprocal interaction of different lncRNA–mRNA pairs has shown a higher number of edges (more than 100) shared by HAND2-AS1 (235 node), MIR100HG (225 node), MAGI2-AS3 (220 node), LINC00578 (179 node), LOC339803 (168 node), LINC02381 (132 node), GAS1RR (120 node) and ADAMTS9-AS2 (104 node) (Additional file 1: Table S4). This high level of connectivity may imply their significant involvement in the process of CRC carcinogenesis (Fig. 3). Ultimately, aligning with the aims of our investigation, we opted to focus on the HAND2-AS1 lncRNA due to its remarkable node connectivity, positioning it as a prime candidate for further downstream analyses.
Fig. 3
The regulatory network of lncRNA–mRNA interactions is constructed by considering significantly differentially expressed genes (DEGs) and DE-lncRNAs. Various colors are employed to distinguish distinct lncRNA molecules, with node sizes varying based on their degree, where larger nodes represent higher degrees. The outer circle encompasses mRNAs with degrees less than 10, while the inner circle represents mRNAs with degrees equal to or greater than 8
Functional and gene set enrichment analysis of HAND2-AS1Functional enrichment analysis was executed by employing the DAVID platform through the clusterProfiler package in R. This facilitated the exploration of noteworthy terms in GO enrichment, encompassing biological process (BP), cellular component (CC), and molecular function (MF), as well as KEGG pathway analyses for both the blue module and HAND2-AS1 targeted DEGs individually. Specifically, for the DEGs within the blue module (Additional file 1: Table S5), the findings revealed prominent processes in the BP category, such as cell-cell signaling, nervous system development, chemical synaptic transmission, regulation of system processes, and regulation of localization, among others (Fig. 4A). In the category of CC, there were significant enrichments in terms like synapse, cell junction, intrinsic component of plasma membrane, extracellular matrix, integral component of plasma membrane, external encapsulating structure, cell projection, etc. (Fig. 4A). Furthermore, terms such as receptor ligand activity, signaling receptor binding, signaling receptor activator activity, and receptor regulator activity emerged as significantly enriched MF categories (Fig. 4A). Additionally, we conducted KEGG pathway enrichment analysis, revealing the top ten enriched terms, including the Calcium signaling pathway, Protein digestion and absorption, cGMP-PKG signaling pathway, IL-17 signaling pathway, cAMP signaling pathway, Cytokine-cytokine receptor interaction, and Wnt signaling pathway (Fig. 4B) (Additional file 1: Table S6).
Fig. 4
GO and KEGG pathway enrichment analyses and GSEA of blue module. A Results illustrating enriched biological processes, cellular components, and molecular functions are provided. The size of each circular node corresponds to the gene ratio of the enriched gene count. B KEGG pathway enrichment analyses reveal the pathways that are enriched. The size of circular nodes reflects the gene ratio of the enriched genes. C Moreover, a Gene Set Enrichment Analysis (GSEA) was conducted on the HAND2-AS1 targeted genes within the TCGA COAD-READ dataset
Furthermore, we conducted distinct GO and KEGG analyses focusing on the HAND2-AS1 targeted DEGs. These analyses revealed enrichment in pathways closely associated with cancer, as illustrated in Fig. 5A, B (Additional file 1: Tables S7, S8). In addition, to gain deeper insights into the biological functions of the HAND2-AS1 targeted DEGs, we employed the GSEA method based on the TCGA COAD-READ dataset. As depicted in Fig. 4C, the targeted DEGs exhibited remarkable enrichment across diverse pathways including adenocarcinoma, adenoma, carcinoma, colon cancer, Crohn’s disease, ulcerative colitis, and inflammatory bowel diseases, all with a significant FDR value of < 0.05.
Fig. 5
The investigation involved Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses, along with Gene Set Enrichment Analysis (GSEA) of genes targeted by HAND2-AS1. A The outcomes encompassed biological process, cellular component, and molecular function analyses. B Additionally, KEGG pathway enrichment analyses were carried out. The size of round nodes corresponded to the gene ratio of the enriched gene number
Expression analysis of HAND2-AS1 in pan-cancer and CRC cell linesWe examined the correlation between HAND2-AS1 expression in cancer tissues and normal tissues using datasets from TCGA and GTEx. Notably, HAND2-AS1 expression was found to be significantly lower in cancer tissues compared to normal tissues in various cancers, including but not limited to breast invasive carcinoma (BRCA), bladder urothelial carcinoma (BLCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), COAD, liver hepatocellular carcinoma (LIHC), ovarian cystadenocarcinoma (OV), acute myeloid leukemia (LAML), pheochromocytoma and paraganglioma (PCPG), READ, stomach adenocarcinoma (STAD), skin cutaneous melanoma (SKCM), testicular germ cell tumors (TGCT), uterine corpus endometrial carcinoma (UCEC), thyroid carcinoma (THCA), and uterine carcinosarcoma (UCS), as depicted in Fig. 6A.
Fig. 6
HAND2-AS1 expression levels exhibit variability across distinct cancer types. A Illustrates HAND2-AS1 level profiles between tumors and normal tissues, focusing on significant cancers. B Depicts the expression levels of HAND2-AS1 in diverse CRC cell lines. *p < 0.05
Additionally, we investigated HAND2-AS1 expression levels across different CRC cell lines. Notably, DLD-1, SW-1463, RKO, and HCT-116 exhibited comparatively higher expression levels, while SW-948, NCI-H747, LS411N, and CL-11 displayed lower expression levels in relation to the other cell lines (Fig. 6B).
HAND2-AS1 prognostic value analysis in pan-cancerWe utilized a univariate Cox regression model to assess the relationship between HAND2-AS1 expression and OS, DFS, PFS, and RFS across diverse cancer types. Remarkably, increased HAND2-AS1 expression was observed to be significantly associated with unfavorable outcomes in various cancers. These included breast cancer, Burkitt lymphoma, colon cancer, CRC, hepatocellular carcinoma, diffuse large B cell lymphoma, lung cancer, lung squamous cell carcinoma, non-small cell lung cancer, ovarian cancer, and pancreatic ductal adenocarcinoma (Fig. 7A). Furthermore, with regard to DFS, higher HAND2-AS1 expression demonstrated a higher DFS rate in breast cancer, colon cancer, CRC, liposarcoma, lung cancer, melanoma, and non-small cell lung cancer (Fig. 7B). Conversely, in the context of PFS, an elevated HAND2-AS1 expression was significantly linked to reduced PFS rates in breast cancer, CRC, diffuse large B cell lymphoma, ovarian cancer, and pancreatic cancer (Fig. 7C). Interestingly, for RFS, elevated HAND2-AS1 expression was significantly associated with decreased PFS in breast cancer, colon cancer, hepatocellular carcinoma, and non-small cell lung cancer (Fig. 7D).
Fig. 7
Forest plot representing univariate Cox regression analysis of HAND2-AS1. A Displays results of univariate Cox regression analysis of HAND2-AS1 for overall survival (OS), B for disease-free survival (DFS), C progression-free survival (PFS), and D for recurrence-free survival (RFS), in various microarray datasets. Red items signify statistical significance
Moreover, survival curves highlighted that lower HAND2-AS1 expression was indicative of notably worse OS (Fig. 8) and RFS (Fig. 9) time in ten and nine distinct cancer types, respectively. Taken together, these findings underscored the potential of HAND2-AS1 as a novel and valuable prognostic biomarker across multiple cancer.
Fig. 8
Kaplan–Meier plots demonstrating the relationship between HAND2-AS1 expression and overall survival (OS) in diverse cancers, highlighting significant outcomes. The pink line signifies high expression groups, while the black line corresponds to the low expression group
Fig. 9
Kaplan–Meier analyses of RFS for HAND2-AS1, utilizing optimal cut-off values across different cancer patient groups. The pink line represents high expression groups, and the black line signifies the low expression group
Immune cell infiltration analysis of HAND2-AS1 in pan-cancerIn order to explore the potential influence of HAND2-AS1 on the response of cancer patients to immunotherapy, a comprehensive analysis was conducted on immune-related data obtained from various algorithms. The analysis was carried out using the ggplot2 package in R. The results consistently revealed a positive correlation between the expression of HAND2-AS1 and the infiltration of various immune components. These immune components included tumor-associated macrophages (TAMs), natural killer (NK) cells, CD4+ T cells, CD8+ T cells, B cells, macrophages, mast cells, dendritic cells, among others. Notably, this correlation was observed across diverse cancers (Fig. 10A). Furthermore, additional data encompassing immune-related pathways, sourced from existing literature, were integrated into the analysis. This integration served to underscore the significant link between the expression of HAND2-AS1 and immune-related pathways. Notable pathways included those associated with the TGF-beta family, interferons, cytokines, and chemokines. These findings, presented in a pan-cancer context (Fig. 10B), collectively suggest that individuals with lower levels of HAND2-AS1 expression might exhibit a relatively immunosuppressive tumor microenvironment. This insight holds potential implications for the design and optimization of immunotherapeutic interventions.
Fig. 10
Explores the interplay between HAND2-AS1 and immune cell infiltration, as well as immune-related pathways. A Depicts the correlation between HAND2-AS1 expression and various immune cells from different algorithms. B Represents the relationship between HAND2-AS1 expression and diverse immune-related pathways
HAND2-AS1 DNA methylation analysis in pan-cancerThe HAND2-AS1 gene methylation data were obtained from the cBioPortal, which showed a substantial increase in the promoter methylation level of HAND2-AS1 in many cancers as depicted in Fig. 11A. Meanwhile, the DNA methylation pattern and HAND2-AS1 mRNA expression were negatively correlated in COAD and READ (Fig. 11B).
Fig. 11
Analysis of HAND2-AS1 DNA methylation data from TCGA. A Illustrates the aggregated methylation value (beta-value) across all samples in the TCGA project. B Presents the correlation between gene expression and methylation value (beta-value) of HAND2-AS1 in TCGA-COAD-READ. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001
Gene alteration of HAND2-AS1 in pan-cancerThe intricate relationship between gene mutations, CNA, and tumor development is well acknowledged. In our investigation into HAND2-AS1 gene alterations, conducted through the cBioPortal platform, a distinct pattern emerged. Notably, the highest frequency of alterations in the HAND2-AS1 gene was observed among patients with ocular melanoma, renal non-clear and clear cell carcinoma. Within this context, the primary alteration type was marked by “mRNA low,“ followed by “mRNA high” alterations (Fig. 12A, D). Furthermore, a comprehensive exploration of the mutation frequencies of HAND2-AS1, characterized by significant deletions and amplifications, was carried out using the GISTIC database. This resource, renowned for its role in dissecting somatic mutations in human cancer, offered valuable insights into the landscape of HAND2-AS1 alterations (Fig. 12B). Across a range of cancers, a prevalent trend of HAND2-AS1 deletions was evident, accompanied by instances of gains and amplifications, albeit to a lesser extent (Fig. 12C). These findings contribute to our understanding of the genetic basis underlying tumor progression and provide potential avenues for further investigation and therapeutic exploration.
Fig. 12
HAND2-AS1 mutation landscape and expression validation. A The relevance of different cancers and HAND2-AS1 expression where it is shown that mutations were mostly relevant to RNA expression. B The putative copy-number alterations from GISTIC of HAND2-AS1 in many TCGA cancers by the cBioPortal database. C Diagram of HAND2-AS1 mutations across cancer types. D mutational profile of HAND2-AS1 across different cancer types. E experimental validation based on 10 tumor and 10 adjacent normal tissues. F, G A set of more external validation based on homogenous GSE87211 (tumor = 202, normal = 157) and GSE68468 (tumor = 186, normal = 55) datasets, respectively
HAND2-AS1 showed significant downregulation in CRC tissuesWith regard to the emerging role of lncRNA HAND2-AS1 as a promising and innovative prognostic, immunomodulatory, and therapeutic biomarker in CRC, we conducted a comprehensive investigation of its expression level in CRC tissues (n = 10) and adjacent normal tissues (n = 10) using RT-qPCR. Our findings, illustrated in Fig. 12E, indicated a significant downregulation of HAND2-AS1 expression in CRC tissues compared to adjacent normal tissues (p = 1e−04). Furthermore, to validate this observation, we performed an external validation using samples from the microarray datasets, GSE87211 and GSE68468. This validation analysis (Fig. 12F, G) consistently demonstrated a substantial under-expression of HAND2-AS1 in CRC samples when compared to normal samples. Thus, in alignment with the TCGA expression analysis, our external validation further strengthened the fact that HAND2-AS1 is significantly downregulated in CRC tissues as opposed to normal tissues.
Comments (0)