
Remember me

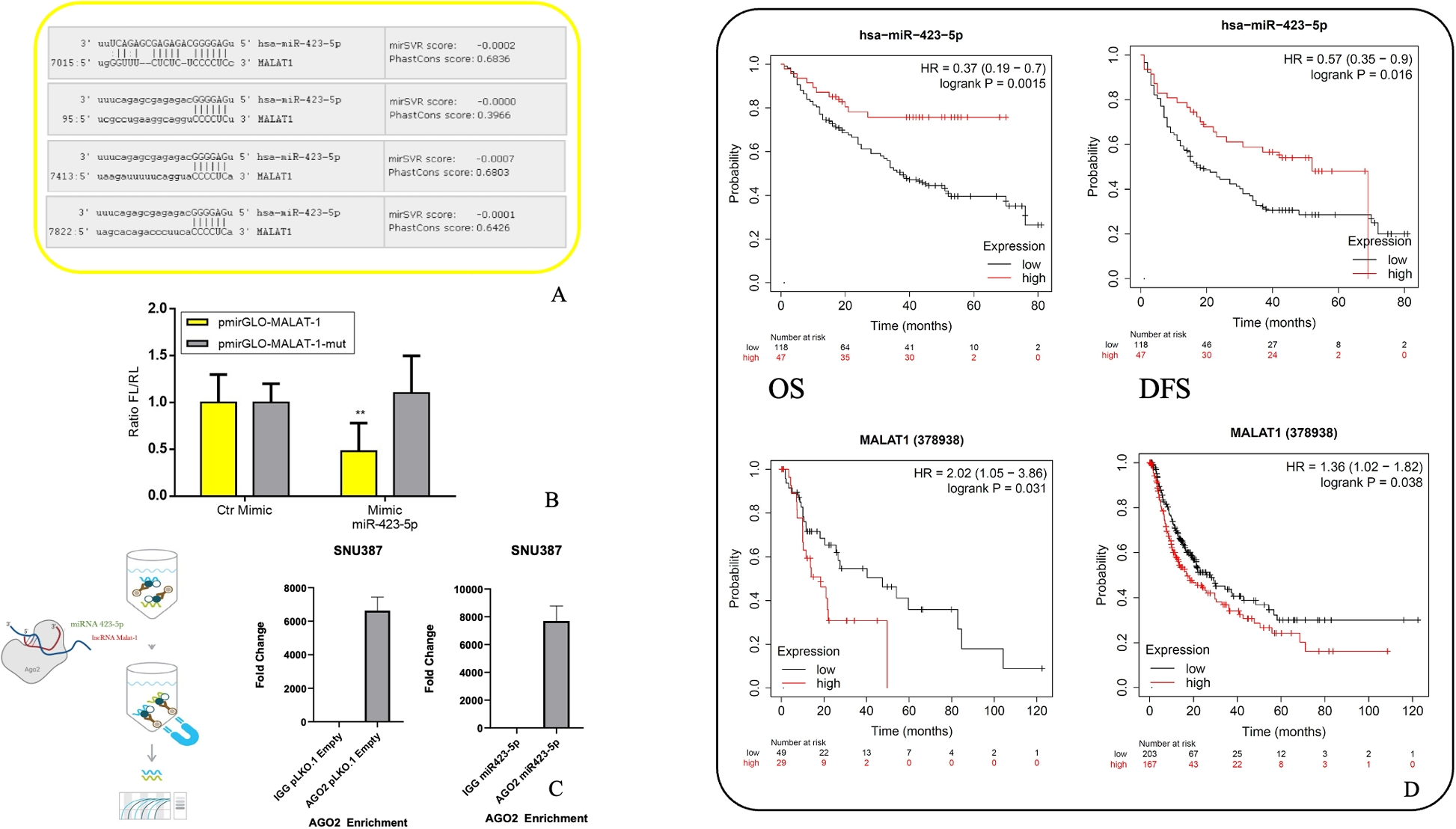



To uncover candidate genes that may play a role in governing stemness in PDAC, we conducted RNA-seq (E-MTAB-3808) on CSC-enriched anchorage-independent sphere cultures in comparison to adherent cultures. We employed a representative selection of five validated PDAC models [21,22,23] to pinpoint genes that exhibit differential regulation within the CSC subset (Fig. 1A, upper panel). Successful enrichment for CSCs was demonstrated by increased expression of stemness-associated genes (Fig. 1A, lower panel). Interestingly, NR5A2 was among the most significantly and consistently upregulated genes (Figs. 1B and S1A). The RNA-seq data could be validated by qPCR using a total of 8 PDX models showing a consistent and significant upregulation of NR5A2 by up to 32-fold in spheres relative to their differentiated counterparts (Fig. 1B). In line with these mRNA data, we found detectable NR5A2 protein levels in differentiated cancer cells. However, NR5A2 expression was further increased in CSCs following anchorage-independent sphere culture or CD133 sorting (Figs. 1C & S1B). These data suggested a specific functional role for NR5A2 in pancreatic CSCs.
Fig. 1
NR5A2 is overexpressed in pancreatic cancer stem cells. A Upregulation of stemness-related mRNAs in CSC-enriched sphere (SPH) versus adherent (ADH) cultures derived from a diverse set of primary PDAC tumors. Representative images of adherent and sphere cultures are shown in the upper panel. The lower panel represents the quantification of mRNA expression. Each sample was analyzed in biological duplicates, which are displayed side by side. The error bars indicate the mean ± standard deviation (SD) from technical duplicates. B RNA sequencing and qPCR validation for NR5A2 expression expressed as fold-change for adherent (ADH) versus sphere (SPH) cultures. The dotted line indicates reference levels for adherent cells, set as 1.0. RNA sequencing data are displayed as pooled data from n = 5 different PDAC cultures using biological duplicates. The qPCR validation was performed from pooled data derived from n = 8 different PDAC cultures. C Western blot analysis for NR5A2 protein levels in adherent (ADH) versus sphere (SPH) cultures in PDX215 and PDX354 cells (upper panel), as well as in CD133 + versus CD133– sorted PDAC cultures (lower panel). β-Actin was used as the loading control. D NR5A2 and NR5A1 mRNA expression in PDAC versus normal tissue as analyzed by GEPIA 2. E Prognostic significance of NR5A2 mRNA expression levels for overall survival (OS) and relapse-free survival (RFS) for PDAC as analyzed by Kaplan–Meier Plotter databases. F UMAP projections of single-cell RNA-seq data consisting of 24 PDAC tumor samples and 11 control pancreases without any treatment, as described by Peng et al. [24]. The UMAP plots illustrate the clustering of PDAC tumor samples into two distinct groups: cancer cells versus normal cells (left, upper panel) and ductal cells versus acinar cells (left, lower panel). Ductal cells were characterized by the expression of KRT19 and AMBP, while acinar cells exhibited high expression of PRSS1. The UMAP plot on the right visualizes the distribution of NR5A2 expression within the ductal and acinar cell clusters. Asterisks indicate significance at the indicated levels: ** p < 0.01, *** p < 0.001. Please also see Supplementary Fig. 1
Notably, using the GEPIA 2 database (http://gepia.cancer-pku.cn/index.html), NR5A2 was found to be most strongly expressed in normal pancreas tissue but downregulated in corresponding PDAC tissue, whereas NR5A1 was not detectable in either tissue (Fig. 1D). NR5A2 mRNA expression was also more strongly expressed in normal liver and bile duct, but no change in expression was observed for the respective cancer tissues (Fig. S1C). All other tissues showed the expected low NR5A2 mRNA expression. When stratifying patients for NR5A2 expression in their respective PDAC tissue, patients with increased NR5A2 expression showed reduced overall survival and a trend towards diminished relapse-free survival (Fig. 1E). Consistent results were obtained using the ArrayExpress database E-MTAB-1791 (Fig. S1D) [25]. Expectedly, no such differences could be observed for other cancer types, such as lung, breast, and bladder cancer (Fig. S1E) due to low NR5A2 expression levels (Fig. S1B). Single-cell RNA-seq of PDAC tissue confirmed that NR5A2 is expressed more prominently in non-transformed acinar and ductal cells in the pancreas compared to transformed PDAC cells (Figs. 1F & S1F) [24, 26]. Notably, however, we could still identify some transformed ductal cells that showed detectable NR5A2 expression, suggesting considerable intratumoral heterogeneity for NR5A2 expression.
NR5A2 regulates proliferation of differentiated PDAC cellsAs NR5A2 expression was rather modest in our differentiated PDAC cells compared to CSCs, we first aimed to validate above findings in our primary PDAC models. For this purpose, we explored a pharmacological inhibitor of NR5A2 (Cpd3) to inhibit the activity of the NR5A2 protein and assessed the treatment effects of Cpd3 in PDAC by tracking NR5A2 mRNA levels. Using three different primary PDAC cultures, Cpd3 showed no acute or unspecific toxicity at 24 h as evidenced by cell viability and toxicity assays (Fig. S2A-B). Still, we observed a substantial reduction in NR5A2 mRNA levels following a single-shot treatment as early as 24 h after drug administration, and the reduction was maintained for at least 72 h (Fig. S2C-D). Consistently, NR5A2 protein levels were also reduced by ~ 50% (Fig. S2E).
Next, we treated differentiated PDAC cells with graded doses of Cpd3 and monitored cell confluency on the Incucyte® platform. Our results showed a decrease in cell confluency with increasing doses of Cpd3 (Figs. 2A & S2F). Notably, Caspase 3/7 immunofluorescence (Fig. 2B & S2G) and AnnexinV flow cytometry (Figs. 2C & S2H-I) revealed no change in the apoptotic rate of the treated PDAC cells after 72 h across the relevant pharmacological range of Cpd3 concentrations. However, we noted a dose-dependent reduction in cell proliferation, as evidenced by flow cytometry using the proliferation marker Ki-67 (Figs. 2D & S2J-K). These findings for pharmacological inhibition of NR5A2 could be further validated using two effective siRNA against NR5A2 (Figs. 2E-F & S2L-M), which corroborated the lack of change in apoptotic cells and the reduction in Ki-67+ proliferative cells after siNR5A2 silencing. In line with these findings, Cpd3 treatment resulted in a marked downregulation of mRNA expression for CCNE2 (G1 cyclin binding CDK2) and upregulation of CDKN1A (cyclin-dependent kinase inhibitor 1; p21), respectively (Figs. 2G & S2N). These changes were accompanied by an increase in CDKN1A (p21) protein levels (Fig. 2H). These results indicate that NR5A2 inhibition specifically affects cell proliferation in differentiated PDAC cells without inducing apoptosis or cell death. Based on these observations, we determined that a Cpd3 dose range of 20-80 μM was suitable for subsequent experiments.
Fig. 2
NR5A2 controls proliferation in differentiated cancer cells and is drugable. A Cell density and morphology were assessed after 72 h of treatment with Cpd3 (40 µM) versus control (Ctrl). Representative results for PDX215 cultures are shown (left). Additionally, the overall cell confluency was monitored over an 80-h period using the IncuCyte® platform (right). B Caspase 3/7 staining for cells treated with control or Cpd3 (40 µM) for 72 h. Quantification and representative images for PDX215 from n = 5 experiments are presented. C Apoptosis analysis was performed using DAPI/Annexin V flow cytometry in PDX215 cells treated with graded doses of Cpd3. The lower right quadrant indicates early apoptosis, while the upper right quadrant represents late apoptosis (marked by the red rectangle). Quantification reveals the percentage of combined early and late apoptotic cells (n = 3). D Flow cytometric dot blot analyses were carried out to examine Ki-67 expression after 72 h of treatment with graded doses of Cpd3 in PDX215 cells. The quantification depicts the percentage of Ki-67+ cells (n = 3). E Apoptosis analysis, measured by DAPI/Annexin V flow cytometry, and the measurement of proliferation (F), indicated by the number of Ki67+ cells, were conducted in PDX215 cells treated with scramble siRNA and the two most effective siNR5A2 variants (#1 and #2) for 72 h. Quantification of four biological replicates is displayed. G qPCR fold change of NR5A2 and CKDN1A (p21) mRNA following 72-h treatment with Cpd3 (80 µM) in PDX215 cells. H Western blot analysis of CDKN1A protein levels following 72 h treatment with Cpd3 (80 µM) in PDX215 cells. Data were presented as mean ± SD and statistically analyzed using two-tailed Mann–Whitney tests. Asterisks indicate significance at the indicated levels: * p < 0.05 and ** p < 0.01. Please also see Supplementary Fig. 2
NR5A2 controls stemness in PDACWe next evaluated the functional effects of NR5A2 inhibition on pancreatic CSCs. First, we treated forming spheres with Cpd3 every 48 h. Cpd3 treatment resulted in a marked inhibition of 3D sphere formation, significantly reducing the number and size of spheres compared to the control (Fig. 3A-B). To further corroborate these data, we treated PDAC cells during the formation of secondary spheres with Cpd3 for 72 h, which showed a sustained decrease in secondary sphere numbers, although this came with a degree of intertumoral variability (Fig. 3C). A consistent decrease in colony numbers on day 21 compared to the control was also noted (Figs. 3D & S3A). These data suggested that Cpd3 significantly reduced the number of CSCs with subsequent loss of sphere and colony-forming capacity. Even after treatment withdrawal, there was no evidence for recovery of the stemness phenotypes. Our findings were further validated using genetic tools for modulating NR5A2 and its effect on secondary sphere formation. Briefly, once the primary sphere formation was finished on day 7, spheres that were larger than 40 μm were collected, dissociated to a single-cell suspension, and then re-cultured for an additional 7-day duration. This process is aimed at further increasing the concentration of cells that display stem-like properties. Intriguingly, knock-down resulted in a consistent decrease in second-generation sphere-forming capacity, whereas NR5A2 overexpression enhanced it (Fig. 3E). Consistent data were obtained for colony formation (Fig. S3B).
Fig. 3
NR5A2 controls stemness in PDAC. A Sphere formation capacity on day 7 following Cpd3 treatment. Representative images for PDX215 and PDX354 following treatment with Cpd3 (60 µM). B Quantification of primary sphere formation capacity on day 7 of Cpd3 treatment for PDX215 (n = 4 biological replicates) and PDX354 (n = 10 biological replicates). Data are shown as violin plots with dotted lines indicating the median. C The schematic (created with biorender.com) illustrates the process of forming first-generation spheres over 7 days. This is succeeded by the disintegration of the formed spheres into a single-cell suspension, and then the commencement of second-generation spheres over another 7-day period, with or without Cpd3 treatment (upper panel). The lower panel shows the secondary sphere formation of cells treated with Cpd3 (60 µM) for five different PDAC cultures. D Colony formation capacity of cells treated with Cpd3 for 72 h. Representative pictures (top), quantification of colony formation (bottom). E Secondary sphere formation capacity following knockdown or overexpression (OE) of NR5A2 for PDX215 and PDX354. In panels C, D, and E data are presented as mean ± SD and statistically analyzed using two-tailed Mann–Whitney tests to compare two groups (n = 4 biological replicates). Asterisks indicate significance at the indicated levels: * p < 0.05, *** p < 0.001, and **** p < 0.0001. Please also see Supplementary Fig. 3
Inhibition of NR5A2 specifically eliminates tumor-initiating CSCsTo further explore a potential bifunctional role of NR5A2 in PDAC, we next investigated its specific role in the CSC subpopulation. For this purpose, we treated second-generation spheres that are highly enriched for CSCs with siNR5A2 for 72 h and closely monitored their CD133+ CSC content. As depicted in Fig. 4A-B, treatment with siNR5A2 resulted in a decrease in CD133+ CSCs. These findings prompted us to investigate whether a similar treatment effect could be observed when targeting NR5A2 with graded doses of Cpd3. Intriguingly, we observed a dose-dependent reduction in the CD133+ CSC population following treatment with Cpd3 (20 to 80 µM) (Figs. 4C-D & S3C). Remarkably, this decline in the CD133+ CSC population was accompanied by a substantial increase in Annexin V staining, indicative of apoptosis specifically within the CD133+ CSC population. This effect could be reproduced by pharmacological inhibition of NR5A2 using Cpd3 treatment and confirmed with siNR5A2 (Figs. 4E-F & S3D-E). Importantly, the differentiated CD133– cancer cells did not exhibit a significant increase in Annexin V staining (Fig. 4E-F), highlighting the specificity of the apoptotic response in the CD133+ CSC population. Notably, PDAC cells following treatment with Cpd3 showed a more pronounced epithelial-like morphology and increased cytokeratin expression (Figs. 4G & S3F). These data demonstrate that NR5A2 has distinct roles in CSCs versus differentiated cancer cells. While NR5A2 drives cell proliferation in differentiated cells, predominantly via downregulation of p21 (CDKN1A) [11], in CSCs, NR5A2 appears to promote stemness and prevent apoptosis.
Fig. 4
Inhibition of NR5A2 specifically eliminates pancreatic cancer stem cells. A Flow cytometry analysis of CD133+ CSCs following 72 h of treatment with siNR5A2 variants #1 and #2; 'src' indicates siScramble. Representative flow cytometry dot plots are displayed. B Quantification of n = 3 biological replicates. C Flow cytometry analysis of CD133+ CSCs following 72 h of treatment with graded doses of Cpd3. Representative data are depicted. D Quantification of CD133+ CSCs in n = 3 biological replicates. E Percentage of apoptotic Annexin V positive cells among CD133– differentiated cancer cells (grey) and CD133+ CSC (blue) following treatment with siNR5A2 variants #1 and #2, and F the percentage among CD133– differentiated cancer cells (grey) and CD133+ CSC (blue) following treatment with 20 and 40 µM Cpd3 in n = 3 biological replicates. G Immunofluorescence for Pan-cytokeratin (green) following 48 h of treatment with 80 µM Cpd3. Nuclei were stained with DAPI (red). H In vivo tumorigenicity of decreasing numbers of highly enriched CD133+ FLUO+ CSCs following pharmacological or genetic targeting of NR5A2. I Flow cytometry for CD133+ CD44+ and CD133+ CXCR4+ CSCs in harvested tumors. In panels A to F, data are presented as mean ± SD and statistically analyzed using two-tailed Mann–Whitney tests to compare two groups (n = 3 biological replicates). In panel I, data are presented as floating bars, and statistically analyzed using two-tailed Mann–Whitney tests to compare two groups (n = 3–6 tumors). Asterisks indicate significance at the indicated levels: * p < 0.05. Please also see Supplementary Fig. 4
In vivo tumorigenicity represents the ultimate test for CSC functionality. Pancreatic CSCs can be enriched to the highest purity through their inherent autofluorescence (FLUO) and the stem cell marker CD133 [7, 9]. Therefore, we sorted CD133+FLUO+ CSCs and pharmacologically or genetically inhibited NR5A2 prior to injecting them into immunocompromised mice. Tumor formation was monitored for six months after injection. As few as ten vehicle-treated CD133+FLUO+ CSCs readily formed tumors, whereas their NR5A2-inhibited counterparts failed to do so (Fig. 4H). Extreme limiting dilution analysis (ELDA) revealed a 45-fold and 49-fold decrease in CSC frequency upon pharmacological or genetic inhibition of NR5A2, respectively. The few tumors that did form after injection of larger numbers of Cpd3-treated cells showed a significant decrease in CSC content, defined as CD133+CD44+ or CD133+CXCR4+ cells (Fig. 4I). These data demonstrated that stemness in PDAC is dependent on NR5A2.
NR5A2 promotes stemness via direct binding to the SOX2 promoter/enhancerTo identify the mechanism by which NR5A2 promotes stemness in PDAC, we screened for the expression of stemness-related transcription factors following treatment with Cpd3 [27, 28]. qPCR was performed after 24 h of NR5A2 inhibition and revealed a significant and reproducible down-regulation of the Sex-determining region Y-BOX-2 (SOX2) transcription factor and a rather unexpected up-regulation of MYC, whereas other stemness-related transcription factors such as NANOG and POU5F1 (OCT-4) remained unchanged (Figs. 5A & S4A-B). These data suggest that NR5A2 might control stemness in pancreatic CSCs by upregulating SOX2 expression and repressing MYC expression through direct or indirect mechanisms.
Fig. 5
NR5A2 promotes stemness via direct binding to the SOX2 promoter/enhancer. A qPCR analysis for mRNA levels of stemness-associated genes following 24 h of treatment with Cpd3 (40 µM, n = 4 biological replicates with four technical replicates). The dotted line indicates baseline expression levels, set as 1.0. B Time course for SOX2 RNA levels following Cpd3 treatment (40 µM) at 24, 48, and 72 h. C Immunofluorescence for SOX2 (yellow) following control (Ctrl) DMSO (top) or Cpd3 (bottom) treatment (40 µM) for 72 h. Nuclei are stained with DAPI (blue). D Western blot for SOX2 protein levels following NR5A2 overexpression (left) or 72 h of treatment with Cpd3 (40 µM) (right). E qPCR analysis of mRNA levels for NR5A2 and SOX2 in response to knockdown of NR5A2 using two different shNR5A2 (sh) in three different PDAC cultures. The dotted line indicates baseline expression levels, set as 1.0. F qPCR analysis of mRNA levels for NR5A2 and SOX2 in response to NR5A2 overexpression (OE) in two different PDAC cultures. G In vivo tumorigenicity of decreasing numbers of the most differentiated CD133–FLUO– pancreatic cancer cells following overexpression (OE) of NR5A2 or SOX2. H Percent input of immunoprecipitated DNA at CDKN1A positive control enhancer, NR5A2 and SOX2 promoters. The intergenic region is used as a negative ChIP control. I CUT&Tag analysis of NR5A2 protein binding at the SOX2 locus. WashU Epigenome browser tracks showing CUT&TAG signals at the SOX2 locus with the indicated transcription start site (TSS). Red signals represent NR5A2 binding in CD133+ PDAC cells (upper track), and purple signals represent NR5A2 binding in CD133- PDAC cells (middle tracks). The black tracks represent the control, consisting of IgG binding in unsorted cells. In panels B, E, F, G, and I, data are presented as mean ± SD and statistically analyzed using two-tailed Mann–Whitney tests to compare two groups (n = 4 biological replicates). Asterisks indicate significance at the indicated levels: * p < 0.05 and **** p < 0.0001
The transcription factor SOX2 is an oncogene strongly upregulated in CSC-enriched spheroid cultures (Fig. S4C). It is a functional driver of CSC phenotypes in PDAC [22, 29]. However, means to modulate SOX2 levels have yet to be described. To determine if SOX2 could be a direct target of NR5A2, we further explored the kinetics of SOX2 downregulation following Cpd3 treatment in several PDAC models. Indeed, Cpd3 substantially decreased SOX2 mRNA levels as early as 24 h, and levels continued to decrease to less than 50% within 72 h (Fig. 5B). Consistently, SOX2 protein levels were significantly reduced by 72 h, as shown by immunofluorescence (Figs. 5C & S4D) and western blot (Fig. 5D). In contrast, other transcription factors such as NANOG, POU5F1, and KLF4 were not altered at 24 h (Fig. S4E). It was not until 72 h that we found their expression levels to be reduced. Therefore, most likely, they occurred secondary to the downregulation of SOX2.
We next used genetic targeting of NR5A2 by lentiviral delivery of shNR5A2 for permanent suppression of NR5A2. We found a consistent decrease in SOX2 mRNA (Fig. 5E), whereas lentiviral overexpression of NR5A2 further upregulated SOX2 mRNA (Fig. 5F) and protein levels (Fig. 5D). Intriguingly, using sorted differentiated CD133–FLUO–cancer cells, overexpression of NR5A2 induced upregulation of CD133 and CXCR4 (Fig. S4F), enhanced sphere formation (Fig. S4G), and, most importantly, markedly enhanced in vivo tumorigenicity (Fig. 5G). Consistent data were obtained for the direct overexpression of SOX2 (Figs. 5G & S4G).
The above data suggested that NR5A2 promotes stemness in PDAC by directly controlling SOX2 expression. To demonstrate such potential direct regulation of SOX2 transcription by NR5A2, we performed a chromatin immunoprecipitation assay for NR5A2 at the promoter of SOX2 and NR5A2 (Fig. 5H). We found an enrichment of NR5A2 binding at both sites relative to the negative intergenic region, comparable to the published binding of NR5A2 at the CDKN1A (p21) enhancer [11]. Notably, this binding was virtually abrogated following treatment with Cpd3. These results demonstrate a direct regulation of SOX2 expression by NR5A2, which can be abolished with Cpd3 treatment. Moreover, analysis of the CUT&TAG signals at the SOX2 locus revealed distinct binding patterns of NR5A2 in CD133+ versus CD133– cells (Figs. 5I & S4H). In CD133+ CSCs, signals indicative of NR5A2 binding were observed directly after the TSS, consistent with NR5A2 binding to the SOX2 promoter region. In contrast, for CD133– cells, signals indicative of NR5A2 binding appeared at a distant site downstream from the TSS. These data suggest that NR5A2 predominantly regulates SOX2 in CD133+ pancreatic cancer stem cells.
NR5A2 regulates CSC metabolism by modulating MYC expressionWe have previously shown that an intricate balance of MYC and PPARGC1A (encoding for PGC-1α) determined the metabolic phenotype of pancreatic CSCs due to their strong reliance on oxidative phosphorylation for maintaining stemness [30]. High MYC expression suppresses PPARGC1A, mitochondrial biogenesis, and oxidative phosphorylation, thereby pushing metabolism towards glycolysis and diminishing stemness. Stimulated by the unexpected upregulation of MYC mRNA expression upon NR5A2 inhibition (Fig. 5A), we hypothesized that this might contribute, at least in part, to the loss of stemness. Therefore, we investigated the MYC/PPARGC1A balance following Cpd3 treatment and found that three different PDAC models showed increased MYC levels while PPARGC1A levels were down-regulated (Fig. 6A).
Fig. 6
NR5A2 promotes stemness by diminishing MYC expression. A qPCR analysis for NR5A2, MYC and PPARGC1A mRNA levels following 72 h of treatment with Cpd3 (40 µM). B Change in oxygen consumption rate (OCR) indicative of mitochondrial respiration following 72 h of treatment with Cpd3 (40 µM) for adherent cultures (Adh, differentiated cancer cells) or sphere cultures (Sph, enriched for CSCs). C Change in extracellular acidification rate (ECAR) indicative of glycolysis following 72 h of treatment with Cpd3 (40 µM) in Adh or Sph cultures. D Change in OCR following overexpression (OE) of NR5A2 in differentiated cancer cells. E Percent input of immuno-precipitated DNA at the MYC promoter following treatment with Cpd3 (40 µM). The intergenic region is used as a negative ChIP control. F OCR levels for shNT or shMYC, following 72 h treatment with DMSO (Ctrl) or Cpd3 (40 µM). G qPCR of mRNA levels for NR5A2, SOX2 and MYC in shNT or shMYC cells, following 72 h of treatment with DMSO (–) or Cpd3 (40 µM). H Changes in maximal (max.) respiration and ATP production in shNT or shMYC cells, following 72 h treatment with DMSO Ctrl (–) or Cpd3 (40 µM). In panels A, E, G, and H data are presented as mean ± SD and statistically analyzed using two-tailed Mann–Whitney tests to compare two groups (n = 4 biological replicates). The asterisk * indicates significance for p < 0.05. Please also see Supplementary Fig. 5
This inverse gene expression change resulted in a drastic switch of the metabolic phenotype of pancreatic CSCs from their preferred oxidative phosphorylation state to a highly glycolytic phenotype (Fig. 6B-C). This switch was associated with a reduced dependency of CSC-enriched spheres on mitochondrial respiration, maximal respiration, and ATP production (Fig. S5A). On the other hand, NR5A2 overexpression in differentiated cancer cells shifted their metabolism toward oxidative phosphorylation (Fig. 6D). To conclusively demonstrate a potential direct regulation of MYC transcription by NR5A2, we performed a chromatin immunoprecipitation assay for NR5A2 at the MYC enhancer. We found an enrichment of NR5A2 binding to the MYC enhancer relative to the negative intergenic region (Fig. 6E). Importantly, treatment with Cpd3 virtually abrogated this binding. These results demonstrate a direct regulation of MYC expression by NR5A2, which can be abolished with Cpd3 treatment.
To determine whether the cellular metabolic phenotype induced by NR5A2 inhibition was indeed functionally mediated by unleashed MYC expression, we targeted MYC expression levels using an inducible shRNA against MYC. As predicted, in untreated PDAC cells, MYC knock-down increased mitochondrial respiration (Fig. 6F), reduced lactate production (Fig. S5B), and subsequently enhanced CSC function, as evidenced by augmented sphere formation capacity (Fig. S5D). These metabolic and phenotypic changes coincided with the upregulation of SOX2 and NR5A2 (Fig. 6G). Intriguingly, the simultaneous knock-down of MYC virtually abrogated the expected metabolic shift toward enhanced glycolysis and glycolytic capacity in response to Cpd3 treatment. Consistently, MYC knock-down also attenuated the increase in lactate production and reversed the impaired sphere formation capacity following Cpd3 treatment (Figs. 6F-H & S5B-D). These data demonstrate that, besides SOX2 activation, inhibition of MYC is an essential mechanism for the stemness-promoting effects of NR5A2, resulting in metabolic reprogramming of the cells toward oxidative phosphorylation.
NR5A2 inhibition targets CSC in vivo and prevents disease relapseThe above data suggest that NR5A2 inhibition could be used as a therapeutic strategy to counteract stemness in PDAC. To demonstrate our findings' potential clinical utility, we performed in vivo intervention studies. In pilot experiments, we used our PDX354 model as an intermediate responder to Cpd3. Due to the lipophilic characteristics of Cpd3, we encapsulated the compound into lipid nanocarriers before i.p. injection. We validated the biological activity of encapsulated Cpd3 (100 mg/kg body weight) by qPCR analysis, showing strong downregulation of NR5A2 and SOX2 with a consistent upregulation of MYC and CDKN1A in vivo (Fig. 7A).
Fig. 7
NR5A2 inhibition targets CSCs in vivo and extends survival in preclinical PDAC models. A qPCR analysis for NR5A2, SOX2, MYC and CDKN1A mRNA levels following 72 h of treatment with Cpd3 (100 mg/kg body weight) in vivo. Data are presented as mean ± SD (n = 4 biological replicates). B Dosing and timing of allocated treatments for tumor-bearing mice. C CSC content following seven days of allocated treatments. CSCs were defined as CD133+ CD44+ cells or CD133+ CXCR4+ cells as assessed by flow cytometry. Data are presented as floating bars with lines indicating the median for n = 3 tumors per group and statistically analyzed using two-tailed t-tests to compare versus control. D Tumor growth in cm3 according to allocated treatments with two treatment cycles of 28 days each as outlined in (B), with n = 8–9 mice per group. Each mouse carried two tumors. Data are presented as mean ± SD. The arrows below depict treatment cycles, rather than specific treatment intervals or durations within each cycle. E Overall survival of tumor-bearing mice following allocated treatment. A total of 18 PDX models were treated using a 2 × 1 × 1 approach (two animals per model per treatment); n = 36 per group. F qPCR analysis of baseline NR5A2 and SOX2 mRNA levels (n = 8–10 biological replicates). Combined gene expression represents the mathematical product of NR5A2 and SOX2 mRNA levels. Data are presented as box and whisker plots with the center line denoting the median value. In panels A, D, and F data are statistically analyzed using two-tailed Mann–Whitney tests to compare two groups. Asterisks indicate significance at the indicated levels: * p < 0.05, ** p < 0.01, and *** p < 0.001. Please also see Supplementary Fig. 6
Next, once PDX354 tumors had reached ~ 0.2cm3 in volume, mice were randomized to vehicle control, Cpd3 alone, gemcitabine alone, or gemcitabine plus Cpd3. The latter was either given early and concomitantly with gemcitabine (during the first and second cycles of chemotherapy) or delayed (only during the second cycle of chemotherapy) (Fig. 7B). We assessed CSC content at the end of the first treatment cycle (day 28). Cpd3 alone significantly reduced the CSC content defined as CD133+CD44+ or CD133+CXCR4+ cancer cells, whereas gemcitabine treatment increased the CSC content (Fig. 7C). The combined simultaneous treatment with gemcitabine and Cpd3 virtually reduced all CSCs to undetectable levels in the tumors.
Notably, the reduced CSC content induced by Cpd3 monotherapy did not translate into significantly reduced tumor growth (Fig. 7D). However, the simultaneous combination of Cpd3 and gemcitabine resulted in instant tumor regression, with no overt relapse over the subsequent 12 weeks. We also tested a successive treatment strategy for gemcitabine and Cpd3 to reduce potential side effects and to avoid the antiproliferative effect of Cpd3, at least during the first cycle of chemotherapy. The successive treatment with delayed Cpd3 treatment still resulted in significant and sustained tumor regression, despite the much larger tumor burden at the time of Cpd3 treatment initiation (Fig.
Comments (0)