Patient information and sample acquisition
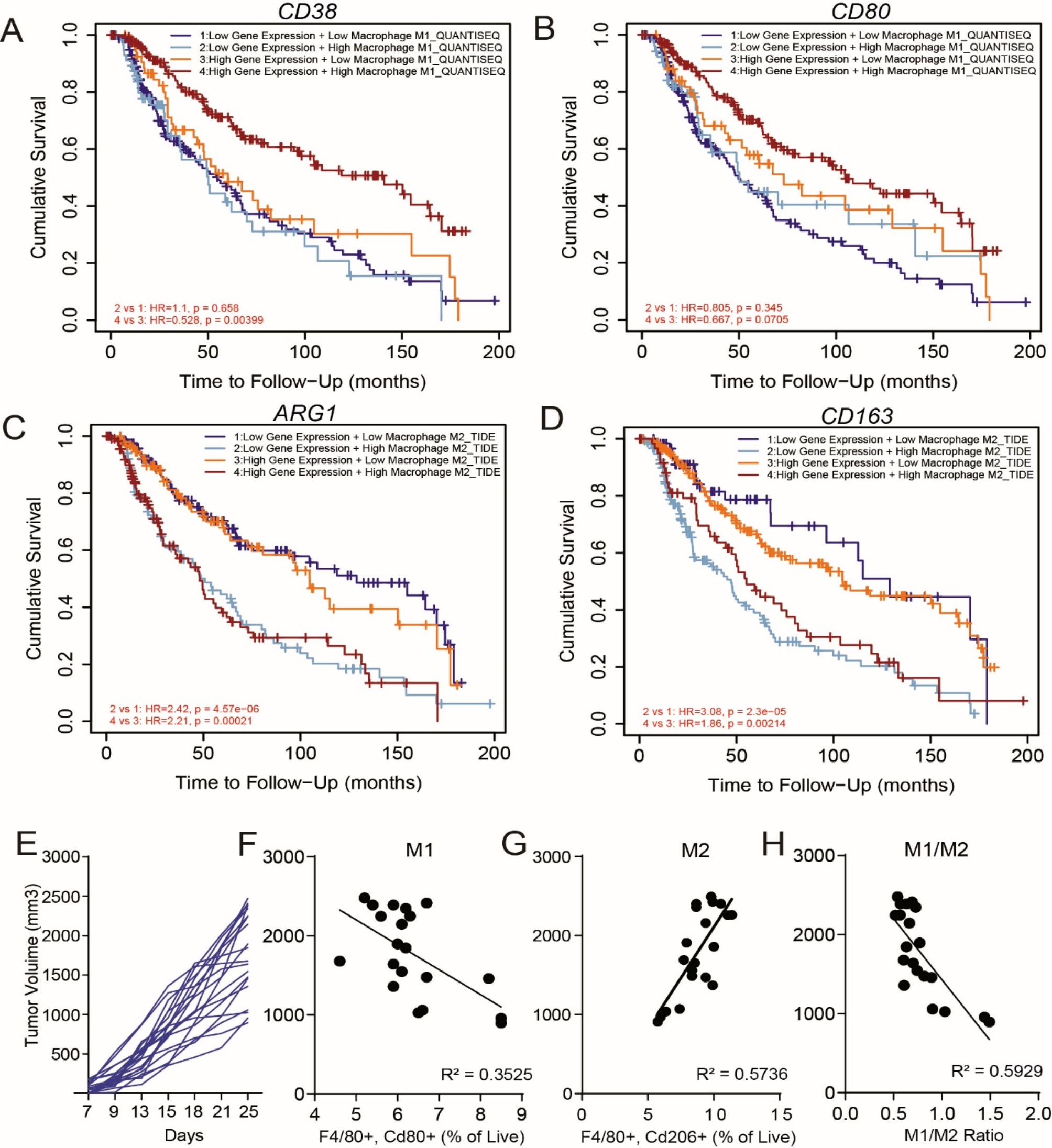
Tissue specimens were gathered from a cohort of 82 advanced pancreatic ductal adenocarcinoma (PDAC) patients who received their primary platinum drug-based chemotherapy treatment at Guangdong Provincial People’s Hospital during 2016–2022. Prior to this initial platinum drug-based chemotherapy regimen, these patients had not undergone any other forms of chemotherapy, radiation therapy, immunotherapy, or targeted therapy. Patients specifically underwent platinum-based chemotherapy following one of the three regimens: mFOLFIRINOX, characterized by an administration of oxaliplatin, irinotecan, leucovorin on the first day and a continuous 46-hour infusion of 5-FU across the first two days, recurring every two weeks; GemOx, entailing an administration of gemcitabine on days 1 and 8, and oxaliplatin on day 1, in cycles of three weeks; or GP, which consisted of gemcitabine on days 1 and 8, and cisplatin on day 1, repeating every three weeks. The therapeutic outcomes were assessed in accordance with the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1. At the four-month mark post the commencement of chemotherapy, patients showing Complete Response (CR) or Partial Response (PR) were categorized as chemosensitive, while those with Stable Disease (SD) or Progressive Disease (PD) were labeled as chemoresistant. progression-free survival (PFS) was ascertained as the duration from the initiation of chemotherapy to the onset of disease progression.
Extraction and cultivation of stromal fibroblasts
Both cancer-associated fibroblasts (CAFs) and primary normal adjacent tissue-associated fibroblasts (NAFs) were extracted from PDAC tissues and corresponding normal tissues. Utilizing the Human Tumor Dissociation Kit (130-095-929, Miltenyi Biotec, Germany), single cells were derived from dissociated samples. The established primary CAFs and NAFs were propagated in a fibroblast medium (FM, ScienCell) that was supplemented with 10% fetal bovine serum, 1% fibroblast growth factors, and 1% penicillin-streptomycin, and maintained at 37 °C in an atmosphere of 5% CO2.
Conditioned medium preparation and co‑culture
Approximately 2 × 106 stably transfected CAFs were propagated in a 10 cm cell culture dish for a duration of 48 h. The medium was subsequently retrieved, centrifuged to eliminate cell debris, and employed to culture Panc-1 and MiaPaCa-2 cells for two weeks, following which they were employed for cytological evaluations.
Cell lines
The PDAC cell lines, Panc-1, MiaPaCa-2 and Bxpc3 procured from the American Type Culture Collection (ATCC), were maintained at 37 °C in a humidified atmosphere containing 5% CO2, employing Dulbecco’s Modified Eagle Medium (DMEM, Gibco) or RPMI-1640 medium (Gibco), supplemented with 10% fetal bovine serum (FBS, Gibco).
RNA isolation and quantitative real‑time PCR (qRT‑PCR)
Total RNA was meticulously extracted from both frozen tissue samples and cultured cell lines with the aid of TRIzol reagent (Life, USA). This RNA was then converted into complementary DNA (cDNA) by employing HiScript Reverse Transcriptase (R101-01, Vazyme, China). The subsequent qRT-PCR analyses were facilitated by the utilization of the CFX96™ Real-Time System (Bio–Rad, USA) in conjunction with a TB Green Premix Ex TaqTM kit (RR820A, Takara, Japan). The relative abundance of the target gene was determined through the 2−ΔΔCt method. The specific primer sequences employed in the qRT-PCR analyses are enumerated in Table S1.
RNase R digestion and actinomycin D assay
In the context of the RNase R digestion assay, total RNA extracted from NAFs and CAFs was treated with or without 5 U/µg of RNase R (RNR07250, Epicenter Technologies), followed by an incubation period of 30 min at 37℃. For the actinomycin D assay, cells were exposed to 2 µg/mL of actinomycin D (Sigma, USA) for designated timepoints extending from 0 to 24 h. Subsequently, qRT-PCR was utilized to ascertain the expression levels of circBIRC6 and BIRC6. Each of these experiments was independently repeated thrice to ensure reliability and reproducibility.
Cell transfection
The complete sequence of circBIRC6 was cloned into the pCD-ciR vector by IGE (Guangzhou, China). siRNA targeting SAE1and EIF4A3 and XRCC4 mutant plasmids were sourced from IGE. The shRNA constructs targeting human circBIRC6 were purchased from IGE. The siRNA and shRNA sequences are included in Table S2.
EVs experiments
EVs were purified from CAFs culture medium as previously described. EVs was derived from the CAFs culture medium, using a pre-established procedure. Briefly, CAFs were cultivated in DMEM, supplemented with EV-free fetal bovine serum (SBI, USA) over a period of 48 h. Post-culture, the medium was collected, subjected to centrifugation at 3000 rpm at 4℃ for 10 min, aiding in the removal of cells and debris. This was succeeded by another centrifugation session at 10,000 × g for 30 min to eliminate microvesicles. After filtering the supernatant using a 0.22-µm filter, it was once again centrifuged at 120,000 × g for 70 min. The resultant pellet, rich in EVs, was rinsed with PBS and subjected to a final round of centrifugation at 120,000 × g for another 70 min. For plasma-derived EVs, we employed an EV isolation kit (Thermo Fisher Scientific, USA). We determined EV characteristics through electron microscopy utilizing negative staining and quantified them using the Nanosight LM10 (Malvern, Framingham, MA) integrated with the NTA v3.1 software (Malvern, Framingham, MA). Primary antibodies against CD9 and TSG101 were harnessed as EV markers.
For the quantification of EV-packaged RNA, we utilized an equal number of EV isolated via ultracentrifugation for RNA extraction and normalization against exogenous λ polyA (Takara, Japan) for the following qRT-PCR analysis.
Pancreatic organoids culture
Freshly harvested human pancreatic cancer tissue was meticulously processed for organoid culture. Initial steps included sectioning the tissue and subjecting it to enzymatic digestion at a constant temperature of 37℃ for increments of 15 min, a cycle that was repeated between 3 and 5 times. The enzyme-digested tissue was then centrifuged to facilitate the collection of the supernatant. Single pancreatic cancer cells, isolated from this procedure, were thoroughly combined with matrix gel. This mixture was subsequently deposited into 24-well plates. Upon solidification of the matrix gel, a culture medium, replete with growth factors, was introduced to support organoid growth.
Western blot
Cells were lysed using RIPA buffer, and the resultant supernatant harvested post-centrifugation. Protein concentration was quantified via a BCA protein assay kit, and equal protein volumes underwent sodium dodecyl sulfate-polyamide gel electrophoresis. Proteins were then transferred to a polyvinylidene fluoride membrane, blocked with a protein-free rapid blocking buffer, and incubated overnight with primary antibodies at 4 °C. Membranes were rinsed in TBST, treated with secondary antibodies for an hour, and immunoreactive bands were visualized with a Chemi XT4 gel imaging system. Used antibodies are cataloged in Table S3.
CCK‑8 assay
PDAC cell drug response was evaluated by seeding 4,000 treated pancreatic cells/well in 96-well plates, followed by oxaliplatin treatment at concentrations ranging from 0 to 1000µM for 72 h. Post-incubation with 10 µl CCK-8 solution (K1018, APExBIO, USA) at 37 °C for 2 h, absorbance at 450 nm was measured using a Tecan microplate reader. Drug response was inferred from the half-maximal inhibitory concentration (IC50) values, computed via GraphPad Prism 8.0. For cell proliferation, cell viability was assessed daily via absorbance readings at 450 nm.
Colony formation assay
700 Panc-1 or MiaPaCa-2 cells underwent designated treatments and were seeded into 6-well plates, left to attach for 24 h, and treated with fresh complete medium supplemented with 4 µM (Panc-1) or 2 µM (MiaPaCa-2) oxaliplatin. After exposed 24 h, the cells were cultured with fresh complete medium for 2 weeks. Colonies were fixed, stained, and manually counted, with three independent repetitions.
Annexin V-PI apoptosis assay
Cell apoptosis was measured using an Annexin V-PI apoptosis detection kit (BMS500FI, Invitrogen, USA). Briefly, the cells were digested by tyrisin without EDTA and Harvested cells were resuspended in binding buffer and stained with FITC-conjugated Annexin V and PI dye for 15 min before flow cytometric analysis within an hour. This assay was performed three times.
RNA sequencing and data analysis
Panc-1 cells at passage 5 were utilized for mRNA profiling via RNA sequencing. Post RNA extraction and cDNA library preparation, sequencing was conducted by Guangzhou Huayin Health Medical Group. Gene set enrichment analysis was executed via GSEA_Linux_4.0.3. For whole-genome transcriptome profiling post 48 h of si-circBIRC6 treatment, RNA extraction, cDNA library prep, and sequencing were carried out by BGI Technology. The clean reads were mapped onto the GRCh38.p13 reference genome using HISAT2 (v2.0.4), and a hypergeometric test-based KEGG enrichment analysis was performed via Phyper.
Neutral comet assay
To assess DNA damage, Panc-1 and MiaPaCa-2 cells were exposed to 50 µM and 30 µM oxaliplatin, respectively, for an hour before harvesting at defined time points. A Comet Assay Kit (Trevigen, USA) was employed to conduct the neutral comet assays, following the guidelines provided by the manufacturer. The samples were subsequently stained using SYBR Gold (Invitrogen, USA) to visualize the DNA. Observation and imaging were carried out using an Olympus FluoView 500 microscope. To quantitatively assess the extent of DNA damage, the tail DNA content was evaluated using the CASP software.
Immunofluorescence
Immunofluorescence was performed as previously described. Briefly, cells grown on confocal dishes were treated with oxaliplatin and then harvested. Post fixation and permeabilization, confocal dishes were treated with antibodies against γH2AX (ab81299, abcam, UK) overnight at 4 °C. The confocal dishes were then rinsed, treated with Alexa Fluor 488-conjugated secondary antibodies, counterstained with DAPI, and visualized via confocal fluorescence microscopy (Carl Zeiss AG, Germany).
pimEJ5-GFP reporter assay
The pimEJ5-GFP reporter assay was executed as previously delineated. Tumor cells underwent transfection with pimEJ5-GFP plasmid using Lipofectamine 3000. Following this, puromycin was utilized to select and establish stable clones. Cells with stable pimEJ5-GFP expression were subsequently transfected with pCBASceI (I-SceI) plasmids using Lipofectamine 3000. After a period of 48–72 h post-transfection, GFP-positive cells were analyzed by Fluorescence Activated Cell Sorting (FACS). The pimEJ5-GFP plasmid was generously provided by Dr. Jeremy Stark (Addgene plasmid #44,026).
Fluorescence in situ hybridization (FISH)
The spatial distribution of circBIRC6 in Cancer-Associated Fibroblasts (CAF) was probed using a FISH Kit (Gene Pharma, Suzhou, China). Cells were fixed, and following the manufacturer’s protocol, were subjected to overnight hybridization with Cy3-labeled circBIRC6 probes at 37 °C. Nuclei staining was carried out with DAPI. Fluorescent signals were captured by confocal fluorescence microscopy (Carl Zeiss AG, Germany). Probe sequences can be found in Table S2.
Subcellular fractionation
Cytoplasmic and nuclear RNA fractions were obtained using a PARIS Kit (Thermo Scientific, USA), according to the provided protocol. Subsequently, the nuclear to cytoplasmic RNA ratio was determined by quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR), with U6 and GAPDH serving as nuclear and cytoplasmic controls, respectively. Primer sequences are available in Table S1. For protein fractions, a subcellular protein fractionation kit (Thermo Scientific, USA) was used following the manufacturer’s protocol, and relative expression of protein in the cytoplasm and nucleus was determined by western blot.
RNA pull-down assays
Biotin-labeled circBIRC6 was purchased from IGE. RNA pull-down assays were conducted using a Magnetic RNA-Protein Pull-Down Kit (Thermo Scientific, USA). In brief, biotinylated circBIRC6, immobilized on streptavidin magnetic beads, was incubated with nuclear extracts at 4 °C overnight. Subsequently, the protein bound to circBIRC6 was eluted and analyzed either by mass spectrometry or western blot. Silver staining was executed using a Pierce Silver Stain Kit (Thermo Scientific, USA), according to the manufacturer’s instructions.
Co-immunoprecipitation (Co-IP)
Co-IP was undertaken using a Pierce Co-IP Kit (Thermo Scientific, USA), adhering to the manufacturer’s guidelines. Briefly, harvested cells were lysed in IP buffer. A solution of 10 µg anti-SAE1/anti-XRCC4 or control IgG was combined with the resin and incubated with a 500 µg protein mixture at 4 °C overnight, while being gently rotated. Following three wash cycles, proteins were extracted for subsequent western blot analysis.
Immunohistochemistry
Immunohistochemistry (IHC) was conducted using paraffin-embedded specimens as previously documented. Briefly, post-deparaffinization, rehydration, and heat-induced antigen retrieval, specimens were incubated with γ-H2AX-specific antibodies overnight at 4 °C. Specimens were then washed and incubated with secondary antibodies, followed by exposure to DAB developer and hematoxylin.
DAB Tunel Cell apoptosis detection
DAB tunel cell apoptosis detection was carried out using a DAB Tunel Cell Apoptosis Detection kit (Servicebio, Wuhan), following the manufacturer’s instructions. Briefly, after deparaffinization, rehydration, Proteinase K digestion, and blocking of endogenous peroxides, specimens were incubated with Equilibration Buffer for 10 min. Specimens were then labeled with TdT incubation buffer (Recombinant TdT enzyme: Biotin-dUTP Labeling Mix: Equilibration Buffer = 1:5:50) for 1 h at 37℃, and subsequently stained with DAB developer and hematoxylin following Streptavidin-HRP reaction.
In situ hybridization (ISH)
ISH was conducted using a double-DIG labeled circBIRC6 probe (IGE, Guangzhou, China) to detect the expression of circBIRC6. Following deparaffinization, rehydration, and proteinase K digestion, the samples were subjected to overnight probe hybridization at 37℃. The hybridization signal was subsequently identified with an Enhanced Sensitive ISH Detection Kit (MK1032, BOSTER, China), as per the manufacturer’s guidelines. Probe sequences are listed in Table S2.
In vivo study
Orthotopic xenograft tumor models were established by introducing 1*10^5 luc-Panc-1 cells per group, which were subsequently treated following the protocols used for subcutaneous models. After 12 days post-implantation, therapeutic interventions were initiated, at which point the first set of in vivo imaging system (IVIS) images was captured. A second round of IVIS imaging was conducted following a 15-day treatment course, after which the tumors were excised. At each point of the IVIS assessment, intraperitoneal injections of D-Luciferin, potassium Salt (150 mg/kg, 40902ES01, Yeasen, China) were administered, facilitating the capture of orthotopic fluorescence images with an in vivo FX PRO system (BRUKER Corporation, USA). Notably, throughout the course of the in vivo study, investigators were blinded to the group assignments during the evaluation of experimental outcomes.
Primary tumor samples procured from oxaliplatin-resistant PDAC patients undergoing surgical procedures were utilized to establish subcutaneous tumors in 4-week-old NOD/SCID/gamma (NSG) mice (F1 generation). Xenografts harvested from these F1 mice were segmented into small sections and subsequently implanted into a second generation of mice (F2). Once these F2 tumors reached a volume of approximately 1500 mm3, they were surgically removed, segmented, and transplanted into a third generation of mice (F3). When the volume of the xenografts reached around 200 mm3, the subjects received a combination treatment regimen of in vivo antisense oligonucleotide (ASO) inhibitors against circBIRC6, the PARP1 inhibitor Olaparib, and oxaliplatin-based chemotherapy. For the delivery of these treatments, in vivo-optimized oxaliplatin (10 mg/kg, intraperitoneal injection), ASO targeting circBIRC6 (5 mg/kg, intravenous injection), or Olaparib (50 mg/kg, intraperitoneal injection) were used as per the experiment’s requirements. Tumor volumes were tracked on a weekly basis and subsequently subjected to further IHC analyses. The evaluation of chemotherapy responses was performed according to human clinical assessment standards.
All animal experiments were executed in strict adherence to the guidelines ratified by the Animal Experimental Research Ethics Committee of South China University of Technology.
Bioinformatic analysis
The secondary structure of circBIRC6 was inferred using the RNAalifold tool (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAalifold.cgi). The binding motifs for XRCC4 were obtained from the POSTAR2 database (http://lulab.life.tsinghua.edu.cn/postar2). The potential SUMO2 binding site on XRCC4 was predicted using the GPS-SUMO bioinformatics tool. Schematic illustration was created with BioRender.com.
Statistical analysis
The animal study incorporated five biological replicates, whereas the remaining experiments employed three biological replicates. Sample sizes were established to provide sufficient statistical power to detect a predefined effect size, predicated on outcomes of initial investigations and pilot experiments. All statistical computations were carried out using SPSS 12.0 software (IBM Corp, USA) and GraphPad Prism version 8.0 (GraphPad Software, USA). Differences between two groups were determined using Student’ s t-test, while one-way analysis of variance (ANOVA) was utilized for comparisons among multiple groups. Survival curves were evaluated using the Kaplan-Meier method complemented by a log-rank test. All error bars denote the mean ± standard deviation (SD). A p-value < 0.05 was considered indicative of statistical significance.





Comments (0)