
記住我
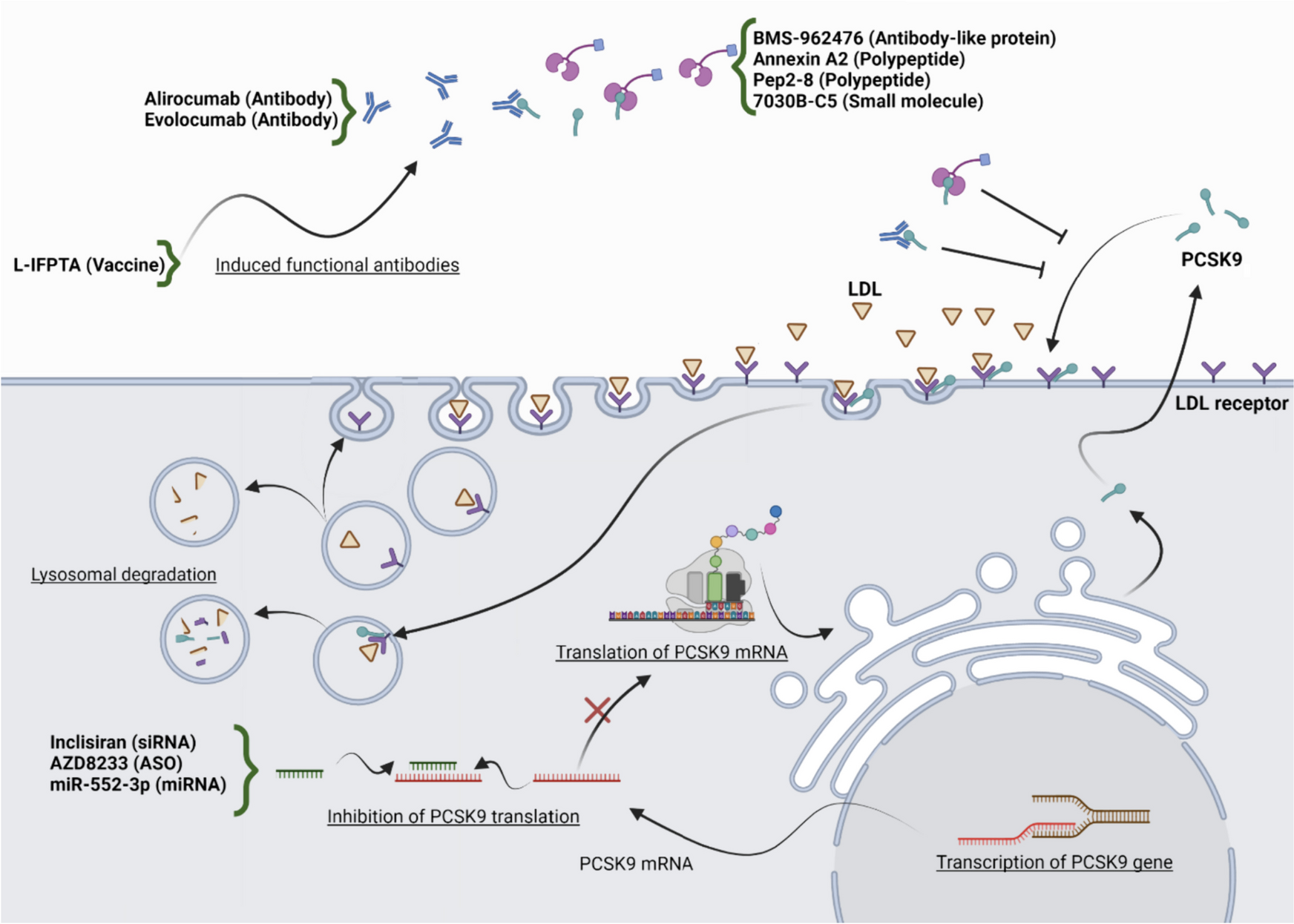





Figure 1 illustrates the flow of this study. Initially, scRNA-seq data pertaining to a single IVDD sample obtained from the GEO database was utilized to elucidate the intrinsic heterogeneity underlying the degenerative process in IVDD. After rigorous quality control procedures, cells exhibiting suboptimal quality were excluded, resulting in the selection of 9,161 cells for subsequent analysis (Fig. 2A). Each cell exhibited a mitochondrial UMI rate below 10%, and a notable correlation was observed between the number of detected genes and sequencing depth. Employing a principled approach, PCA-based dimensionality reduction was performed with a resolution value set to 1, identifying a total of 18 distinct cell clusters. These clusters exhibited pronounced heterogeneity, reflecting diverse cellular states within the intervertebral disc. Leveraging marker genes reported in prior studies, comprehensive cell type annotation was performed, and six categories of cells were characterized, including NPCs, nucleus pulposus progenitor cells (NPPC), macrophages, notochord cells (Noto), neutrophils, and endothelial cells (ECs) (Fig. 2B). Furthermore, proportional analysis of different cell types in the samples indicated that NPCs occupied more than 90% of the NP tissue, while the proportion of Noto in severe IVDD was lower compared to that in mild IVDD (Fig. 2C).
Fig. 1 Fig. 2
Fig. 2
Enrichment analysis of hub genes in IVDD. A Following standard quality control on all cells pertaining to mild and severe IVDD, 9,161 cells were included in the analysis. B A UMAP plot illustrates six distinct cell types within the dataset, as identified through unsupervised clustering (NPCs, nucleus pulposus cells; NPPCs, nucleus pulposus progenitor cells; Noto, notochord cells; EC, erythroid cells). Each color represents a specific cell type. C Proportions of different cell types in mild-IVDD and severe-IVDD samples. D UMAP plots depict the distribution of AA metabolic activity in each cell. E, F A violin plot displays the AA metabolic score of various cell types in mild IVDD and severe IVDD. G, H A violin plot displays the AA metabolic score of NPCs in mild IVDD and severe IVDD
Dynamic modulation of AA metabolism during NPC degenerationSignificant variations were observed in AA metabolic pathway activity across various stages of IVDD, with a positive correlation observed between higher degenerative degrees and increased AA metabolic pathway activity (Fig. 2D). Additionally, among all cell types examined, NPCs exhibited the most pronounced fluctuations in AA metabolic pathway activity, demonstrating elevated levels in cells characterized by greater degeneration (Fig. 2E, F). Therefore, NPCs were further investigated in this study. Subsequently, transcriptomic data pertaining to NPCs were extracted, and a reanalysis was conducted employing UMAP visualization (Fig. 2G). To assess AA metabolic pathway activity in each cell cluster of NPCs, the "AddModuleScore" function was utilized. The results are presented in Fig. 2H. Cluster 3 displayed the highest activity, while Cluster 0 exhibited the lowest activity.
Cell communication analysis of NPCs exhibiting varying AA metabolic activity: unveiling the intricate interplayTo elucidate the impact of AA on the microenvironment of the intervertebral disc, the differences in intercellular communication across different cell types were analyzed utilizing the “CellChat” R package. NPCs were stratified into three distinct categories based on quartiles (25% and 75%) derived from prior assessments; specifically, NPCs were categorized as exhibiting high, medium, or low AA metabolic activity. The construction of the intercellular communication network involved the aggregation of interaction frequencies and their corresponding weights. Figure 3A and B provide a visual representation of the strength of interactions in both incoming and outgoing signal pathways, thus emphasizing the central and intricate role that NPCs play in intercellular communication. Notably, NPCs with high AA metabolic activity exhibited elevated signal strength in outgoing pathways compared to the other two categories of NPCs, whereas NPCs with low AA metabolic activity exhibited heightened activity in incoming signal pathways (Fig. 3C, D). Moreover, a subsequent analysis, focused on ligand–receptor (LR) pairs, delved into the reciprocal communication patterns between NPCs of the aforementioned three categories and other cell types. NPCs with high AA metabolic activity demonstrated the ability to engage in cellular communication with NPPCs through ANGPTL4–(ITGA5 + ITGB1), ANGPTL4–CDH11, ANGPTL4–SDC2, FGF2–FGR1, and PDGFC–PDGFRA interactions; with EC through ANGPTL4–(ITGA5 + ITGB1) and ANGPTL4–CDH5 interactions; and with Noto via ANGPTL4–(ITGA5 + ITGB1), ANGPTL4–SDC2, ANGPTL4–SDC3, ANGPTL4–SDC4, FGF2–FGFR1, NAMPT–(ITGA5 + ITGB1), and NAMPT–INSR interactions. Additionally, NPCs with low AA metabolic activity engaged in cellular communication with NPPCs through FGF7–FGFR1, POSTN–(ITGAV + IGTB5), and PROS–AXL interactions; with Noto via ANGPTL2–(ITGA5 + ITGB1), FGF7–FGFR1, FGF7–FGFR2, GAS6–AXL, GRN–SORT1, POSTN–(ITGAV + ITGB5), and PROS1–AXL interactions; with macrophages through FGF7–FGFR1, GRN–SORT1, POSTN–(ITGAV + ITGB5), and SEMA3E–PLXND1 interactions; and with ECs through FGF7–FGFR1, POSTN–(ITGAV + IGTB5), PROS–AXL, and SEMA3E–PLXND1 interactions (Fig. 3E). These findings highlight the potential significance of AA metabolic pathway activity in NPCs for their communication with various cell types through these receptor interactions.
Fig. 3
Diagram illustrating the intercellular communication network among NPCs exhibiting varying AA metabolic activity and other co-localized cell types. A, B Diagram depicting interactions between NPCs of varying AA metabolic activity and other cell types. The thickness of the connecting lines between two cell types indicates their interaction weight or intensity. C Point plots of outgoing and incoming signal pathways in various cell types. D Heatmap depicting the intensity of intercellular interactions among eight distinct cell type© (E). Summary of LR interactions between NPCs of varying AA metabolic activity and other cell types. The horizontal axis represents ligand cells and their corresponding receptor cells, while the vertical axis represents different signaling pairs
Analyzing the interactions between NPCs and other cell types via LR analysis revealed that these interactions were closely associated with several cellular processes, including cell proliferation, apoptosis, oxidative stress, ECM formation, and lectin response. This study revealed that within the TGFβ signaling pathway, NPCs of diverse AA metabolic types function as receptors that are influenced to varying degrees by macrophages (Fig. 4A). In signaling pathways linked to cell proliferation, NPCs of distinct AA metabolic types serve multiple roles as recipients, mediators, and influencers. They are also influenced by other cell types, with particularly strong associations observed between NPCs exhibiting low AA metabolism (Fig. 4B, C). Regarding processes involving the ECM, fibroblast differentiation signaling pathways, and lectin response signaling pathways, NPCs characterized by low AA metabolism assume more active roles as influencers (Fig. 4D–F). In contrast, within stress signaling pathways, NPCs with high AA metabolism function were observed to be more potent mediators (Fig. 4G–I). Therefore, it is evident that NPCs with different AA metabolic types play pivotal roles in regulating the onset and progression of IVDD by modulating the immune microenvironment.
Fig. 4
Circos diagram illustrates the interrelationships between NPCs exhibiting varying AA metabolic activity and other cell types across various signaling pathways. A heatmap showcases the relative likelihood of NPCs exhibiting varying AA metabolic types playing four distinct roles (sender, receiver, mediator, and influencer) within the signaling pathways. The color intensity corresponds to the magnitude of cellular impact
Pseudotime analysis revealed alterations in NPC metabolic activity in response to varying AA metabolic activityTo further investigate the cellular trajectories of distinct clusters of NPCs involved in AA metabolism, the Monocle pseudotime algorithm for pseudotime analysis was employed, as shown in Fig. 5A, where various NPC clusters are distinguished by different colors. In this pseudotime analysis, shades of blue denote the temporal progression of cell differentiation, with darker shades signifying earlier stages of differentiation. The results presented in Fig. 5B demonstrate that NPCs underwent differentiation from right to left over time, with the lightest shade of blue corresponding to the most recently differentiated cell state 1, while cell states 2 and 3 represent later stages of NPC differentiation. Figure 5C illustrates the distribution of early-stage degenerated NPCs and late-stage degenerated NPCs during the differentiation process, while Fig. 5D showcases the distribution of distinct subgroups of NPCs along the cellular differentiation trajectory. By examining the variations in AA metabolic activity among these subgroups, Cluster 1, characterized by the lowest metabolic activity, was determined to primarily reside in the early stages of the cellular trajectory, corresponding to early degenerated NP tissue. Conversely, Cluster 3, exhibiting the highest metabolic activity, predominantly occupied the later stages of the cellular trajectory, indicative of late-stage degenerated NP tissue. These findings robustly support the close association between AA metabolic activity and IVDD progression.
Fig. 5
Pseudotime trajectory analysis of NPC clusters. A Pseudotime trajectory differentiation plot of NPCs. B Pseudotime trajectory depicting distinct differentiation states of ©s. C Differentiation of NPCs in mild IVDD and severe IVDD. D Pseudotime trajectory differentiation plot based on different clusters of NPCs
Screening hub genes via machine learning algorithmsThree well-validated machine learning algorithms, namely LASSO, SVM-RFE, and RF, were employed to identify essential biomarkers associated with IVDD in datasets GSE70362 and GSE176205. The LASSO algorithm was subjected to rigorous tenfold cross-validation to ensure the robustness of the findings. On the basis of the optimal lambda value (0.07000546), the most influential features were judiciously selected to construct the LASSO model, leading to the identification of nine pivotal genes (PLA2G1B, PLA2G4F, PLB1, PTGDS, AKR1C3, ALOX5, CYP2B6, EPHX2, and ALOX15) (Fig. 6A). Additionally, the RF approach identified 12 significant genes (AKR1C3, ALOX15, ALOX5, CYP2B6, EPHX2, LTA4H, PLA2G1B, PLA2G4F, PLB1, PTGDS, PTGES2, and PTGES3) (Fig. 6B, C). Employing the SVM-RFE algorithm, the classifier was observed to achieve the highest accuracy and a commendable AUC value when employing a set of 29 optimal feature genes. Noteworthy among these genes were PTGDS, AKR1C3, PLB1, ALOX5, ALOX15, EPHX2, PLA2G1B, GGT5, CYP2E1, ALOX12B, PTGES2, PLA2G4F, CYP2B6, CBR1, TBXAS1, LTC4S, PLA2G12A, GPX3, GPX7, LTA4H, PTGS1, PLA2G6, ALOX15B, PLA2G5, CBR3, PTGES3, CYP2C19, PTGES, and PLA2G3 (Fig. 6D, E). After obtaining the aforementioned machine learning results, the R package was utilized to generate plots depicting five hub genes (AKR1C3, ALOX5, CYP2B6, EPHX2, and PLB1) based on machine learning-filtered hub genes and differentially expressed genes (DEGs) (Fig. 6F). Figure 6G also demonstrates that individual hub genes exhibited precision in distinguishing different stages of IVDD (all predicted probabilities were greater than 0.7). Figures 6H–L depict the differential expression of these five hub genes in the NP tissue of IVDD. Among them, AKR1C3, CYP2B6, and PLB1 showed high expression in severe IVDD, whereas the opposite expression pattern was observed for the other genes.
Fig. 6
Development of the early diagnosis model for IVDD. A ROC curves of SVM and RF models. B The impact of the number of decision trees on the error rate. The x-axis represents the number of decision trees, and the y-axis indicates the error rate. The error rate stabilized at approximately 500 decision trees. C Results of the RF classifier's Gini coefficient approach. Genetic variation is on the x-axis, and the significance index is on the y-axis. D, E Gene selection process using SVM-RFE and tenfold cross-validation in the GSE70362 and GSE176205 datasets. The highest model accuracy was achieved when 29 genes were selected. F UpSet plot showcasing the characteristic genes in LASSO, RF, DEGs, and SVM-RFE. G ROC curve values for the five hub genes in the GSE70362 and GSE176205 datasets. H–L Representative bar graphs reveal the expression differences in ALOX5, AKR1C3, CYP2B6, EPHX2, and PLB1 between mild IVDD (n = 17) and severe IVDD (n = 16)
Development of the AI modelTo validate the classification potential of the hub genes, an ANN was initially utilized to construct an AI prediction model using the GSE70362 and GSE176205 datasets. The primary objective was to demonstrate the predictive capability of the hub genes for the early diagnosis of IVDD. In the R "neuralnet" package, the ANN architecture comprised five input layers, five hidden layers, and two output layers. Specifically, through the computation of the gene weights, optimal discrimination between mild and severe disc degeneration was achieved (Fig. 7A). Subsequently, ROC curves were generated to evaluate prediction accuracy. The AUC values approached 1 (AUC = 0.961), indicating the model's robustness. To assess the diagnostic model's performance, validation was performed using the test dataset (GSE67567), which yielded an AUC of 0.720. The AUC across all datasets, including both training (GSE70362 and GSE176205) and validation (GSE67567) datasets, amounted to 0.852. Furthermore, as depicted in Fig. 7B–D, the diagnostic efficacy of the AI model in discriminating between mild IVDD and severe IVDD substantially outperformed that of C-reactive protein (CRP) [33]. Thus, AI models exhibited significant diagnostic value within the entire dataset, enabling more precise assessment of inflammation levels and effective differentiation between various stages of IVDD.
Fig. 7
Development of the early diagnosis AI model for IVDD. A. The AI-ANN structure consists of five convolution layers, three max-pooling layers, and two fully connected layers. B AUC validation in the GSE70362 and GSE176205 datasets. C AUC validation in the GSE67567 dataset. D AUC validation in all datasets (GSE70362, GSE176205, and GSE67567)
IVDD classification based on AA-related hub genesTo further investigate the relationship between disc degeneration and hub genes, an analysis of the expression profiles of hub genes in 43 IVDD samples was conducted using k-means unsupervised clustering. The most stable number of subtypes was determined to be two (k = 2) (Fig. 8A). Subsequently, based on the CDF curves, the 43 IVDD samples were clustered into two distinct subgroups: Subtype A (n = 15) and Subtype B (n = 28) (Fig. 8B, C). These findings align with those of the ANN model, indicating that a higher proportion of patients with Subtype A exhibited severe disc degeneration, whereas those classified as Subtype B tended to display milder disc degeneration, as represented in the Sankey diagram (Fig. 8C).
Fig. 8
Identification of two different IVDD subtypes based on five hub genes. A Unsupervised clustering based on hub gene expression and consensus matrices for k = 2. B Overall CDF curves. C A bar chart illustrates the proportions of mild-IVDD and severe-IVDD groups in the two subtypes. D–I GSEA of Subtype A and Subtype B
Pathway activity and immune infiltration landscape between two stages of IVDDTo elucidate the functional implications of the hub genes and the underlying mechanisms influencing the progression of IVDD, GSEA was conducted to identify dysregulated biological processes and pathways between the two subgroups. The aim was to determine which biological processes were substantially enriched in one group compared to the other. The results revealed that Subtype B exhibited enrichment in the following processes: cellular response to external stimuli, reactive nutrient deprivation, reactive signaling interleukins (ILs), reactive cell dynamics signaling systems, and reactive metabolic disease signaling (Fig. 8D–H). Conversely, Subtype A showed higher richness in amino acid and derivative metabolism (Fig. 8I). To further investigate potential differences in relevant functions and pathways between these distinct subtypes, analyses were performed using the KEGG, Reactome, and WikiPathways databases to reveal differences in the underlying mechanisms of disease progression between the subtypes. Compared to Subtype B, Subtype A exhibited a remarkable upregulation of inflammatory-related pathways, including apoptosis, the prostaglandin metabolic pathway, pantothenic acid and CoA biosynthesis, the peroxisome pathway, oxidative phosphorylation, as well as the metabolism of nitric oxide and glutathione (Fig. 9A).
Fig. 9
Biological characteristics of different IVDD subtypes. A A representative heatmap depicting the differences in the immune landscape in IVDD between Subtype A and Subtype B, as determined using CIBERSORT, TIMER, QUANTISEQ, CIBERSORT-ABS, EPIC, and XCELL algorithms. The bar chart on the right elucidates the correlation between immune cells or stromal cells and their respective subtypes. B A heatmap illustrating differential pathway activity between the two subtypes, based on GSVA using the KEGG, Reactome, and WikiPathways databases
Single-cell analysis showed different interactions between NPCs with varying AA metabolic activities and the immune microenvironment. Therefore, it was imperative to investigate the correlation between these subtypes and immune cell populations. To this end, various algorithms, including CIBERSORT, TIMER, QUANTISEQ, CIBERSORT-ABS, EPIC, and XCELL, were employed to assess immune cell infiltration levels across the subtypes. Subtype A exhibited a heightened infiltration of pro-inflammatory immune cells, including M1 macrophages, whereas Subtype B displayed elevated levels of anti-inflammatory immune cell infiltration, including myeloid dendritic cells, CD4 + T cells, and endothelial cells (Fig. 9B).
Prediction of subtype-specific small molecular compounds and their mechanisms of actionSubtype A- and Subtype B-specific small molecular compounds were predicted utilizing the XSum algorithm to evaluate their potential as drug candidates against the distinct subtypes. Based on the disease subtyping approach based on the five hub genes, XSum suggested several drugs for the treatment of IVDD, namely AH6809, TTNPB, MS-275, NU1025, and clofibrate. The chemical structures of these drugs are illustrated in Fig. 10A–F.
Fig. 10
Five small-molecule drugs identified through XSum analyses. A–F PubChem database displays the molecular structures of the five targeted drugs, including AH6809 (B), TTNPB (C), MS-275 (D), NU-1025 (E), and clofibrate (F). G–L Visualization of molecular docking
Drug–gene interaction and molecular docking analyses of hub genesBased on the XSum results, molecular docking analysis of AH6809 with AKR1C3, CYP2B6, EPHX2, ALOX5, and PDE1B revealed robust docking of AH6809 with these hub genes (Fig. 10G). The binding affinity between AKR1C3 and AH6809 was -7.7 kcal/mol. In this interaction, the hydroxyl group of TYR at position 24 in AKR1C3 formed a hydrogen bond with the carbonyl oxygen atom of AH6809 at a distance of 2.9 Å. Similarly, the carbonyl group of LEU at position 54 in AKR1C3 formed a hydrogen bond with the hydroxyl group of AH6809 at a distance of 2.5 Å. Additionally, the phenyl ring of TRP at position 227 in AKR1C3 engaged in π–π conjugation with the phenyl ring of AH6809 at a distance of 4.4 Å (Fig. 10H). Supplementary file S2 presents the docking results for AH6809 with the other hub genes (Fig. 10I−L).
Friends analysis of hub genesUsing the Friends algorithm, AKR1C3 was found to hold the greatest biological significance among the five hub genes (Fig. 11A). As depicted in Fig. 11C, several pro-inflammatory pathways were significantly negatively associated with AKR1C3, including the IL1R, oxidative stress pathway, pentose phosphate metabolic pathway, and chemotaxis behavior of macrophages.
Fig. 11
Inhibiting AKR1C3 using AH6809 treats IVDD. A A box plot illustrates the functional similarity of the five hub genes, as revealed through Friends analysis. AKR1C3 exhibits the highest degree of correlation with other genes. B AKR1C3 expression in NPCs transfected with an NC negative control (empty plasmid) or an AKR1C3 overexpression plasmid, as detected by RT-qPCR. Data are expressed as mean ± SD (n = 3) (*, P < 0.05; **, P < 0.01; ***, P < 0.001). C Correlation between AKR1C3 and various biological processes. D, E Apoptosis in NPCs transfected with a control NC plasmid or an AKR1C3 overexpression plasmid, as detected by flow cytometry. Data are expressed as mean ± SD (n = 3) (*, P < 0.05; **, P < 0.01; ***, P < 0.001). F Changes in gene expression of anabolic markers (ACAN and COL2A1) and catabolic markers (MMP-3, ADAMTS5, and COX2), as determined by RT-qPCR. Data are expressed as mean ± SD (n = 3) (*, P < 0.05; **, P < 0.01; ***, P < 0.001)
AH6809 alleviates IVDD by inhibiting AKR1C3To date, AKR1C3 has not been reported in association with IVDD. To corroborate the biological alterations resulting from the upregulation of AKR1C3, qRT-PCR was employed to assess the relative efficacy of AKR1C3 overexpression in NPCs (Fig. 11B). To further substantiate this conjecture, overexpression of AKR1C3 was induced in the presence of TBHP. The results indicated that AKR1C3 overexpression enhanced TBHP-induced apoptosis in NPCs (Fig. 11D, E). Subsequent experiments revealed that AKR1C3 overexpression influences NPC proliferation and apoptosis, ECM synthesis and degradation, as well as the release of inflammatory factors by NPCs. RT-qPCR demonstrated an increase in catabolic markers, including MMP-3 and ADAMTS5, coupled with a decrease in anabolic markers, such as ACAN and COL2A1, as AKR1C3 expression levels increased (Fig. 11F). Furthermore, AKR1C3 was found to impact IVDD by modulating the synthesis and metabolism of cyclooxygenase-2 (COX2). These findings collectively establish that AKR1C3, as an integral component of the hub gene network, actively participates in the regulation of IVDD.
Furthermore, given the pronounced affinity between AKR1C3 and AH6809, efforts were initiated to validate the connection between AH6809 and AKR1C3. The results revealed that AH6809 possesses the capacity to alleviate IVDD by suppressing AKR1C3 expression, subsequently influencing apoptosis in NPCs and ECM synthesis. In summary, these results imply that AH6809 holds promise as a potential treatment for IVDD through the inhibition of AKR1C3.
留言 (0)