Rats
We purchased ten-week-old male Wistar rats (body weight, about 300 g) from Sankyo Labo Service (Tokyo, Japan) and maintained them under specific pathogen-free conditions at animal facilities accredited by the Keio University Institutional Animal Care and Use Committee. All rats were kept under a 12-h light/dark cycle and were fed standard diets. Animal experiments were carried out in accordance with Guidelines and Institutional Guidelines on Animal Experimentation at Keio University.
ASCL-PLCs
We established ASCLs from adipose-derived mesenchymal stem/stromal cells (ASCs) as previously described [11, 12, 14]. ASCLs represent a more homogeneous population than ASCs and fulfill criteria for mesenchymal stem cells set by the International Society for Cellular Therapy. Cultivation of ASCLs in the presence of MK lineage induction medium led to peak production of platelets within 12 days. ASCL-PLCs or PRPs (each 1.0 × 107 cells) suspended in 50 µl phosphate-buffered saline (PBS), or PBS alone were activated with 10 mM CaCl2 and incubated 15 min. In this condition, clots did not form in either ASCL-PLCs or PRPs. Both preparations were then frozen at – 80 ℃ and thawed at room temperature immediately before use.
Rat Achilles tendonitis model
Tendonitis models were generated under general anesthesia in 10-week-old male rats (n₌48). Control rats underwent sham-surgery via a skin incision in the right hind limb. Model rats received a similarly-sized skin incision on the posterolateral side of the left limb, and the Achilles tendon and flexor tendon wrapped in the paratenon were identified. The paratenon was opened and the Achilles tendon was cut at 3 mm away from the calcaneus without a gap using a scalpel. After the skin was sutured with 5–0 monofilament nylon, either ASCL-PLCs (1.0 × 107 cells/50 µl) or PBS (50 µl) were injected into the left Achilles tendon site using a 27-gauge needle. Bilateral Achilles tendons were harvested three days after surgery for real-time PCR (n₌24), and at 1 (n₌12), 2 (n₌6) or 4 weeks (n₌6) later for histopathology or fluorescent immunohistochemical analysis. All methods were carried out in accordance with the ARRIVE guidelines.
PRP
To obtain PRP, 30 ml of peripheral blood was collected from healthy adult human volunteers, with approval from the Ethics Committee of our institute (Approval No. 20210093), and blood sampling was carried out with patient consent. Blood was collected into a tube containing citric acid. PRP was obtained by centrifugation (200 × g) at room temperature for 10 min [15], and the PRP (leukocyte-poor; final platelet counts 1.0 × 107/50 µl) was activated with CaCl2 and stored at – 80 ℃. All patients provided written informed consent. The study was approved by the Keio University Institutional ethics committee (Approval No. 20210093).
NIH3T3 cell culture procedures
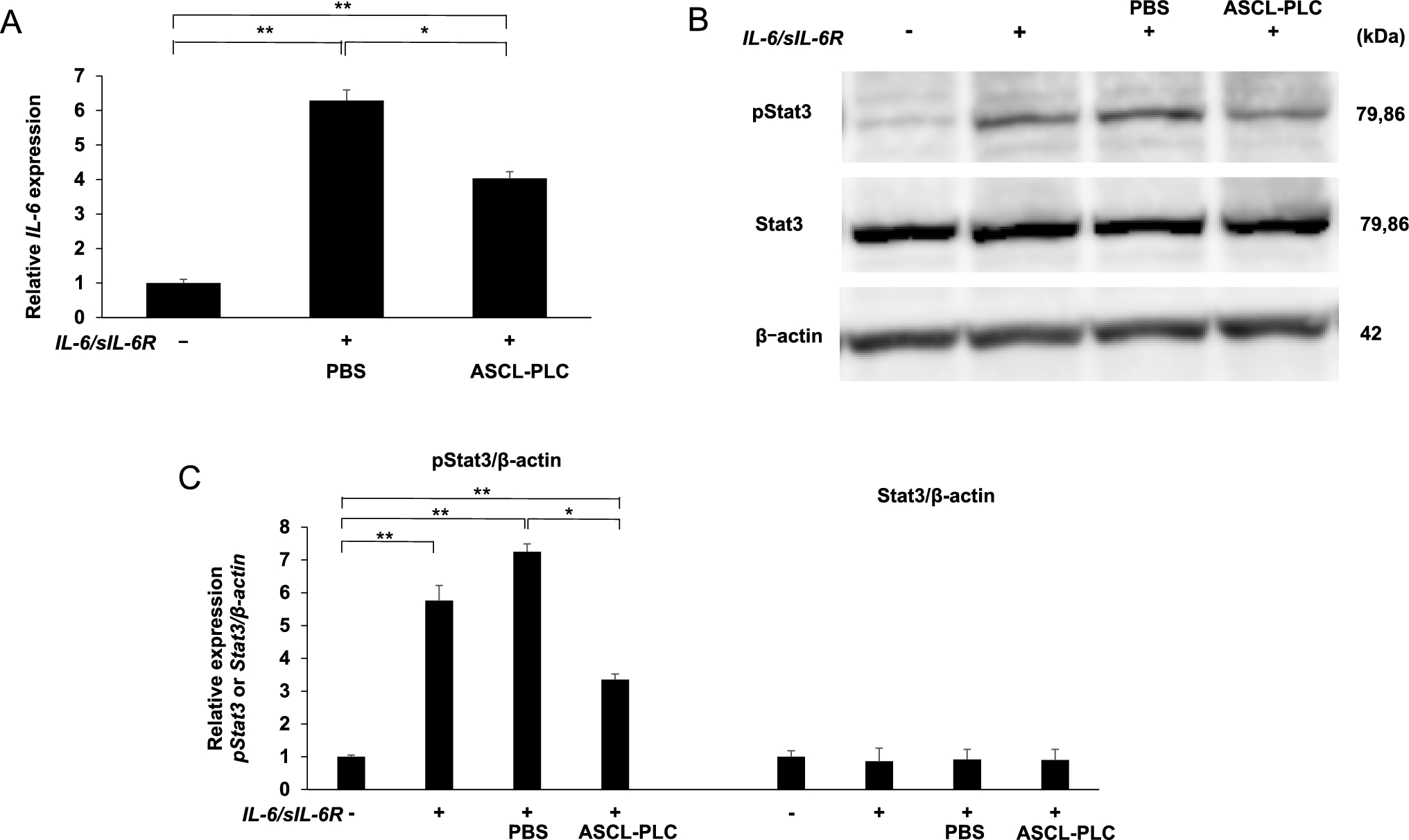
Adherent NIH3T3 fibroblastic cells, which were maintained in DMEM (Sigma–Aldrich Co.) plus 10% fetal bovine serum (FBS) with penicillin G/streptomycin, were cultured in 96-well plates (1.0 × 105 cells/well) with ASCL-PLC (1.0 × 107 cells/well) with or without both IL-6 (100 ng/ml, R & D Systems) and soluble IL-6 receptor (sIL6-R, 100 ng/ml, R & D Systems). After six hours of cultivation, total RNA was collected to assess IL-6 transcript levels. We also cultured NIH3T3 cells in 96-well plates (1.0 × 105 cells/well) with or without ASCL-PLC (1.0 × 105 cells/well) in the presence or absence of various MAPK inhibitors (2 µM anti-p38, 8 µM anti-JNK or 2 µM anti-ERK) and with or without both IL-6 (100 ng/ml, R & D Systems) and sIL-6R (100 ng/ml, R & D Systems). After six hours of cultivation, mRNA was collected for analysis.
Realtime PCR analysis
At the time of surgery, the Achilles tendon was cut at 3 mm from the calcaneal attachment site. Three days later, we identified that region under a stereomicroscope, cut out the Achilles tendon with a scalpel, and minced it into small pieces with scissors. RNA was then extracted from those tissues using Trizol reagent (Molecular Research Center, Inc., Cincinnati, OH). Total RNA was similarly prepared from mouse NIH3T3 cell lines described above. In all cases, single-stranded complementary DNAs (cDNAs) were synthesized with reverse transcriptase (FUJIFILM Wako Pure Chemical Corp., Osaka, Japan). Realtime PCR was performed using SYBR Premix ExTaq II (Takara Bio Inc., Otsu, Shiga, Japan) with a DICE Thermal Cycler (Takara Bio Inc.), according to the manufacturer’s instructions. β-actin expression served as an internal control. Primer sequences for real-time PCR were as follows:
β-actin (rat)-forward: 5’-TCCTCCCTGGAGAAGAGCTATG-3’.
β-actin (rat)-reverse:5’-TGCCACAGGATTCCATACCCAG-3’.
IL-6 (rat)-forward:5’-TCTCTCCGCAAGAGACTTCCA-3’.
IL-6 (rat)-reverse: 5’-GGTCTGTTGTGGGTGGTATCC-3’.
IL-1β (rat)-forward: 5’-TGTGATGTTCCCATTAGAC-3’.
IL-1β (rat)-reverse: 5’-AATACCACTTGTTGGCTTA-3’.
β-actin (mouse)-forward: 5’ -TCCTCCCTGGAGAAGAGCTATG-3’.
β-actin (mouse)-reverse: 5’-TGCCACAGGATTCCATACCCAG-3’.
IL-6 (mouse)-forward: 5’-GTCCTTAGCCACTCCTTCTG-3’.
IL-6 (mouse)-reverse: 5’ -CAAAGCCAGAGTCCTTCAGAG-3’.
Histopathology and fluorescent immunohistochemical analysis
Ankle joints including Achilles tendons were removed one, two or four weeks after ASCL-PLC or PBS injection, fixed in 10% neutral-buffered formalin, embedded in paraffin blocks, and cut into 4-μm sections. Ankles were decalcified in 10% EDTA, pH 7.4, before embedding. Hematoxylin and Eosin (HE) staining was performed according to standard procedures. Cells were then observed using a BioRevo microscope and corresponding software (Keyence, Tokyo, Japan). For fluorescent immunohistochemistry assay, after deparaffinization, sections were microwaved 10 min in 10 mM citrate buffer solution (pH 6.0) for antigen retrieval. After blocking one hour with 3% BSA in PBS, sections were stained for 18 h with rabbit anti-mouse IL-6 (1:100 dilution; Abcam) at 4 °C. After PBS washing, sections were stained for one hour with Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:100 dilution; Invitrogen) at room temperature. DAPI (1:1000; (FUJIFILM Wako Pure Chemical Corp., Osaka, Japan) served as a nuclear stain, and cells were observed under a fluorescence microscope (Keyence, Tokyo, Japan).
Immunoblotting analysis
Whole-cell lysates were prepared from cell cultures using RIPA buffer (1% Tween 20, 0.1% SDS, 150 mM NaCl, 10 mM Tris–HCl (pH 7.4), 0.25 mM phenylmethylsulfonylfluoride, 10 µg /mL aprotinin, 10 µg/mL leupeptin, 1 mM Na3VO4, 5 mM NaF (Sigma-Aldrich Co.)). Equivalent amounts of proteins were separated by SDS-PAGE and transferred to a PVDF (Polyvinylidene Difluoride) membrane (EMD Millipore Corp, Burlington, MA, USA). Membranes were blocked in buffer-containing 10 mM Tris–HCl (pH 7.4), 150 mM NaCl, 0.1% Tween 20, and 5% bovine serum albumin, and incubated with each primary antibody overnight at 4 °C. Primary antibodies used were anti-STAT3 (#4904), anti-pSTAT3 (#9131), (Cell Signaling Technology, Inc., Danvers, MA, USA) or anti-actin antibody (Sigma-Aldrich Co., St Louis, MO, USA). Secondary antibodies were goat anti-mouse IgG (G21040, Thermo Fisher Scientific, Waltham, Massachusetts, USA) and goat anti-rabbit IgG (G21234, Thermo Fisher Scientific). Membranes were then incubated using the appropriate secondary antibodies, and the immune complexes visualized using the ECL Western Blotting Analysis System (GE Healthcare, Tokyo, Japan). Image J v. 1.51 (the National Institutes of Health, Bethesda, MD) was used to quantify each band.
Statistical analysis
Results are expressed as means ± s.d. Statistical significance of differences between groups was evaluated using unpaired two-tailed Student’s t-test or ANOVA († P < 0.10; * P < 0.05; ** P < 0.01; *** P < 0.001; NS, not significant, throughout the paper).







Comments (0)