Somatic reversion, a phenomenon observed in various primary immunodeficiencies, significantly modifies the clinical outcomes of these pathologies [4]. It can be positive, neutral or negative, depending on the gene. Interestingly, despite the detection of immune cell subsets expressing functional proteins and exhibiting restored functionality, somatic reversion improved clinical outcomes only in certain conditions [4, 18]. For instance, it has a positive clinical impact on ADA deficiency, Wiskott-Aldrich syndrome (WAS), Fanconi anemia, and in variants affecting DOCK8, ITGB2, or CXCR4 [18]. However, it negatively impacts patients with somatic variants in CARD11, RAG1, as well as in a reported case of IL2RG reversion in tissue infiltrating T-cells, all associated with Omenn syndrome [4, 18]. Additionally, one patient with a reversion in NEMO, developed refractory inflammatory colitis, as his revertant T cells activated NF-kB in response to growth signals and had a growth advantage over cells carrying the germline change [19]. In other cases, somatic reversion can be neutral, as seen in CD247 or Ligase IV [20] deficiencies, where WT reversions were unable to modify the clinical or immunological phenotype. It has been hypothesized that somatic revertant variants are common in proliferative tissues, like the hematopoietic system, but are limited by the need for functional protein restoration [21, 22].
In the case of CD247 deficiency, only five patients with homozygous germline variants have been reported. Among these, four exhibited a small population of revertant T cells (Table S1), but this did not correlate with improved clinical outcomes as all experienced life-threatening infections, with only one surviving post-hematopoietic stem-cell transplantation (HSCT).
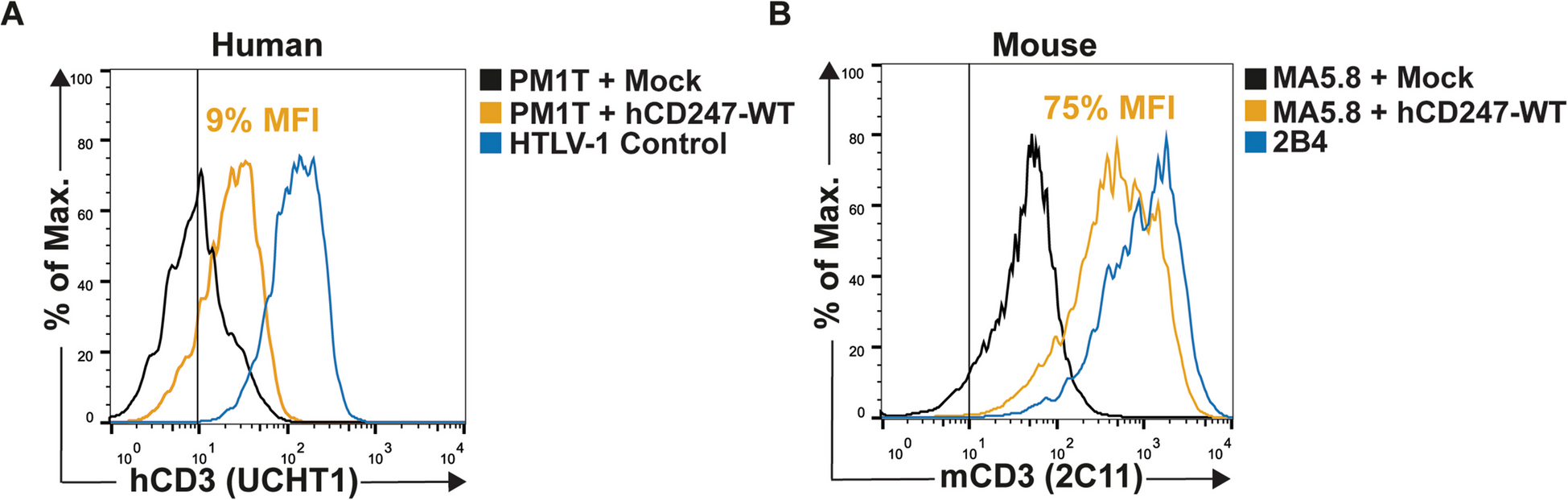
Since no functional studies have explored the potential effect of CD247 somatic reversions, one main objective of our study was to understand, at the molecular level, the impact of different variants on TCR surface expression and CD247-dependent T-cell functions. To this end, we chose the main CD247 somatic reversions (WT, p.Q70W, p.Q70L, and p.Q70Y) in comparison to the germline change (p.M1T, and p.Q70X) reported by Marin [13] and Rieux-Laucat [17], respectively. We found varying degrees of surface TCR expression restoration among the revertants (WT > Q70L > Q70W > Q70Y > > Q70X), which was consistent across different staining antibodies. As expected, the p.Q70X germline variant showed no restoration (Fig. 4A, B and Table S2). This suggests that the revertants can partially or fully restore TCR expression, whereas the germline variant is incapable of doing so. These differences might indicate that the specific amino acid changes in each revertant differentially affect the efficiency of TCR complex assembly and surface expression.
Notably, by testing murine and human cell lines side-by-side, we learned that CD247-deficient mouse T cells MA5.8 cannot be used to model human CD247 deficiencies, since Q70X, which is expectedly unable to restore TCR expression in human CD247-deficient T cells (PM1T or ZKO), does so in MA5.8 (Fig. 2 and Table S2). Therefore, previous reports using such murine cell lines should be reinterpreted in light of our findings [5, 23]. A potential limitation of this conclusion is that they are drawn from a truncated protein (Q70X) that is translated upon transfection and transduction, respectively, from a cDNA, and thus may not exist in primary patient’s T cells due to nonsense-mediated RNA decay (NMD), which is prevented when using cDNA. Rieux-Laucat et al., reported in the discussion that CD247 Q70X was detected in small amounts in the cytoplasm, but not on the membrane in primary patient’s T cells [17]. Although no data were included to support that contention, impaired Q70X protein expression could be partly due to NMD. We identified a CD247 Y152X nonsense variant [14, and unpublished results] in primary patient’s T cells, which was seemingly unaffected by NMD, likely due to its location in the last exon of the gene. In contrast, the reported c.301C > T (p.Gln101X) variant, which is closer to the Q70 codon, results in a premature translation-termination codon (PTC) predicted to cause a truncation of the encoded protein or absence of the protein due to NMD (Variation ID: 419227). Together, these findings confirm the impact of PTC location in NMD and suggest that the Q70X variant could be an NMD candidate. Genome-edited cell lines with Q70X are in progress to address this and other questions in more detail in the future.
Interestingly, analysis of the impact of revertant variants on TCR function, by measuring surface expression of CD69 and CD25 after stimulation with SEE-loaded Raji cells, revealed patterns of CD69 upregulation (Q70W > WT > Q70L > Q70Y > > Q70X) (Fig. 4C, left) and CD25 + cells (Q70W > WT > Q70Y = Q70X > Q70L) (Fig. 4C, right) which did not correlate with surface TCR expression. The p.Q70W variant showed the highest functional restoration, while the p.Q70L variant, despite good TCR expression recovery, was less effective in inducing CD25 expression upon TCR engagement. These results indicate that the p.Q70W variant, although not the best at restoring TCR expression, is the most effective in triggering T cell activation, as shown by higher TCR-mediated CD69 upregulation. We hypothesize that Q70W outperforms wild-type (WT) and Q70L in CD69 and CD25 upregulation due to its aromatic ring, which may facilitate stronger interactions with signaling partners, enhancing ITAM phosphorylation and activation. In contrast, Q70L hydrophobic nature likely hinders essential hydrogen bonding, impairing ITAM accessibility and phosphorylation. This lack of interactions may impair early/intermediate activation events, including CD69 upregulation and sustained CD25 induction. This further suggests that certain CD247 reversions may enhance signaling pathways downstream of the TCR, leading to more robust T-cell activation.
In silico predictors PolyPhen, SIFT or Saphetor classified Q70X as strongly pathogenic, Q70W and Q70Y as probably damaging, while Q70L is considered benign. Therefore, the expected clinical hierarchy based on these predictors would be (WT = Q70L > Q70W = Q70Y > > Q70X). Our phenotypic results generally confirm but also refine such predictions (WT > Q70L > Q70W > Q70Y > > Q70X). The functional results add further layers of complexity, as expression is required for function: Q70W > WT > Q70L > Q70Y > > Q70X (by CD69 upregulation), Q70W > WT > Q70Y = Q70X > Q70L (by CD25+ cells) and WT > > Q70L = Q70W = Q70Y > > Q70X (by Zap70 phosphorylation, as reported by Rieux-Laucat [17]). We thus believe that our cellular model to interrogate variants is more informative than purely in silico predictors.
In addition, the discrepancy between the TCR expression levels and the degree of TCR activation, suggests that Q70 substitutions impact CD247 function beyond surface expression. Q70, a polar residue, likely stabilizes CD247 via hydrogen bonding. Replacing it with hydrophobic (leucine and tryptophan) or amphipathic (tyrosine) residues may alter conformation and signaling. Q70L supports TCR expression but weakens activation, likely due to the loss of hydrogen bonding crucial for ITAM phosphorylation. Q70W disrupts surface expression but enhances signaling, possibly by facilitating stronger ITAM interactions through its aromatic ring. Q70Y impairs both expression and early activation, likely because its bulky aromatic ring and polar hydroxyl group disrupt the structural integrity or proper folding of CD247. However, its hydroxyl group may allow limited interactions that support moderate CD25 expression, indicating partial preservation of sustained signaling pathways.
Given the crucial role of CD247 in TCR selection and tolerance, the clinical implications of the results obtained with the Q70W and Q70L variants are significant. The Q70W heightened induction of CD69 and CD25 could potentially lead to excessive T-cell activation, disrupting the immune balance and increasing the risk of autoimmune diseases and chronic inflammation. Conversely, the Q70L variant, with its impaired T-cell activation, may result in immunodeficiency, increasing susceptibility to infections and related complications.
In addition, none of the CD247 revertants or the germline change induced ZAP-70 phosphorylation after TCR stimulation with an anti-CD3 mAb (Fig. S7 and Table S2). This can be attributed to the fact that ZAP-70 phosphorylation is an immediate (very early) event during T cell activation, whilst CD69 and CD25 upregulation are early events, relatively distal from TCR signaling initiation (which peak at 24 and 48 h post-stimulation, respectively); thus, allowing the cell to respond to cumulative TCR signaling from CD247 somatic variants, or, alternatively, from CD3 receptor subunits [24].
Overall, these findings highlight that restoring TCR expression does not always lead to functional recovery, and each revertant shows a unique restoration profile, underlining the importance of evaluating multiple functional markers to fully understand the impact of somatic reversions. However, we are fully aware that for missense variants, overexpression from strong promoters can compensate for the functional defects of the protein, so it would be advisable for this kind of experiments, to generate cell lines that express the variants of interest from the endogenous gene and not generating knock-out cell lines that are transduced with a cDNA that is overexpressed.
Concerning the discordant effects on expression and function of the studied variants, Kaiser et al. [21] reported a CD247-deficient patient (Table S1) with a frameshift change in the CD247 leader peptide, exhibiting more than 30 non-WT somatic variants that could, to varying degrees, restore surface TCR expression. Some of them persisted for months and some others did not, irrespectively of surface TCR expression levels, suggesting the existence of discordant effects on expression and function (measured as T cell survival in vivo), although such effects were not operating in TCR assembly interactions, but rather in leader peptide functions. In other genes, some examples have been reported, too. Reconstitution assays in vitro with pre-TCRα variants found in patients showed that some restored surface pre-TCRα expression partially but were fully impaired functionally [25]. Similarly, expression of a NEMO variant protein was not markedly reduced by flow cytometry, but its activity was defective, as confirmed by an NF-κB luciferase reporter assay [19].
In conclusion, our results indicate that somatic mosaicism in CD247 is a common, random event in T cells. However, its capacity to restore TCR expression does not match TCR function. Additionally, none of the tested revertants, including WT, improved the patients’ survival, likely because these events took place too late in T cell development to have a clinical impact, as suggested by Kaiser [21] and Attardi [22]. These findings may be relevant to understand the role of CD247 in TCR structure and function during human T cell development in vivo and its impact on human immunodeficiencies.




Comments (0)