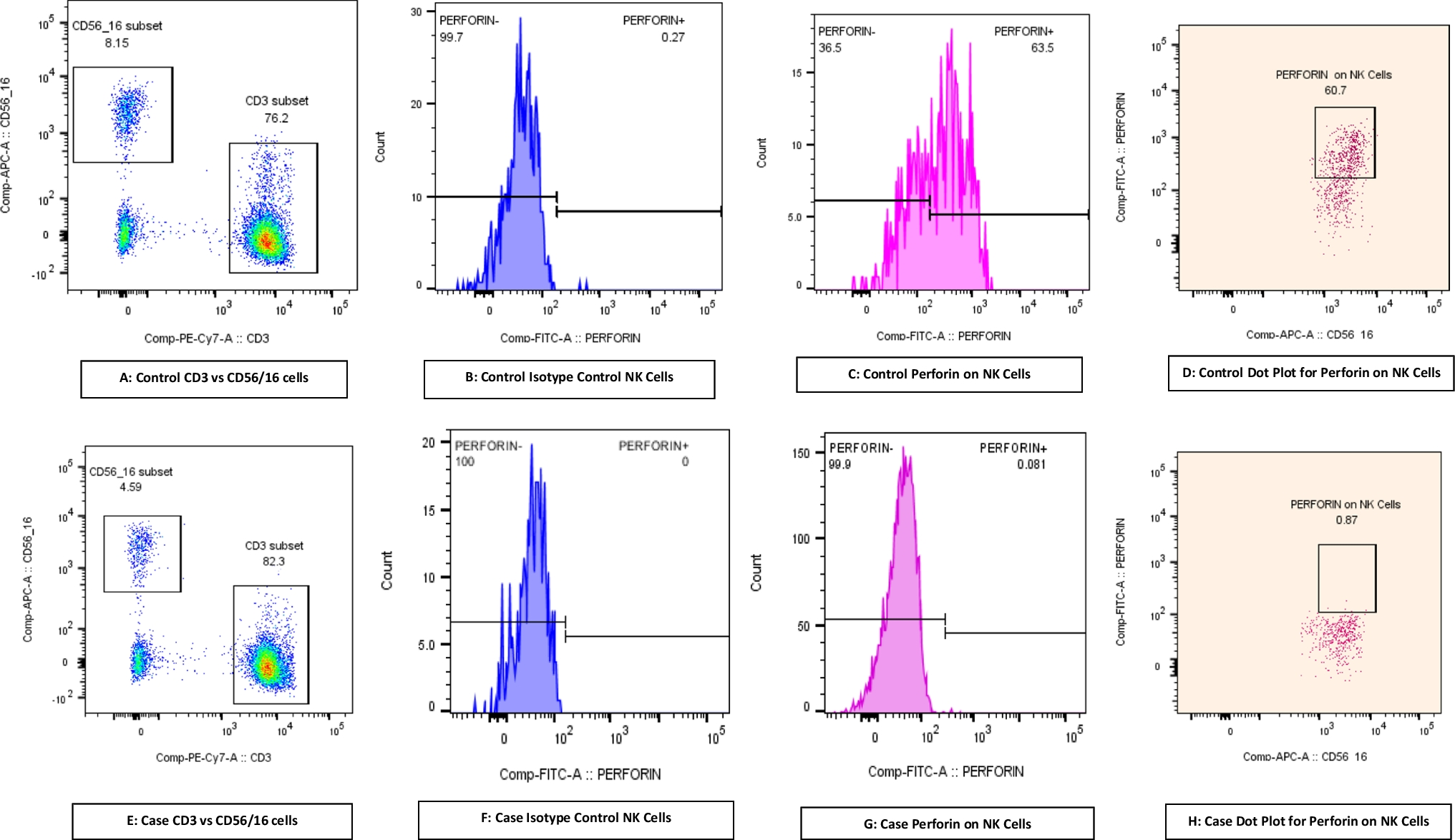
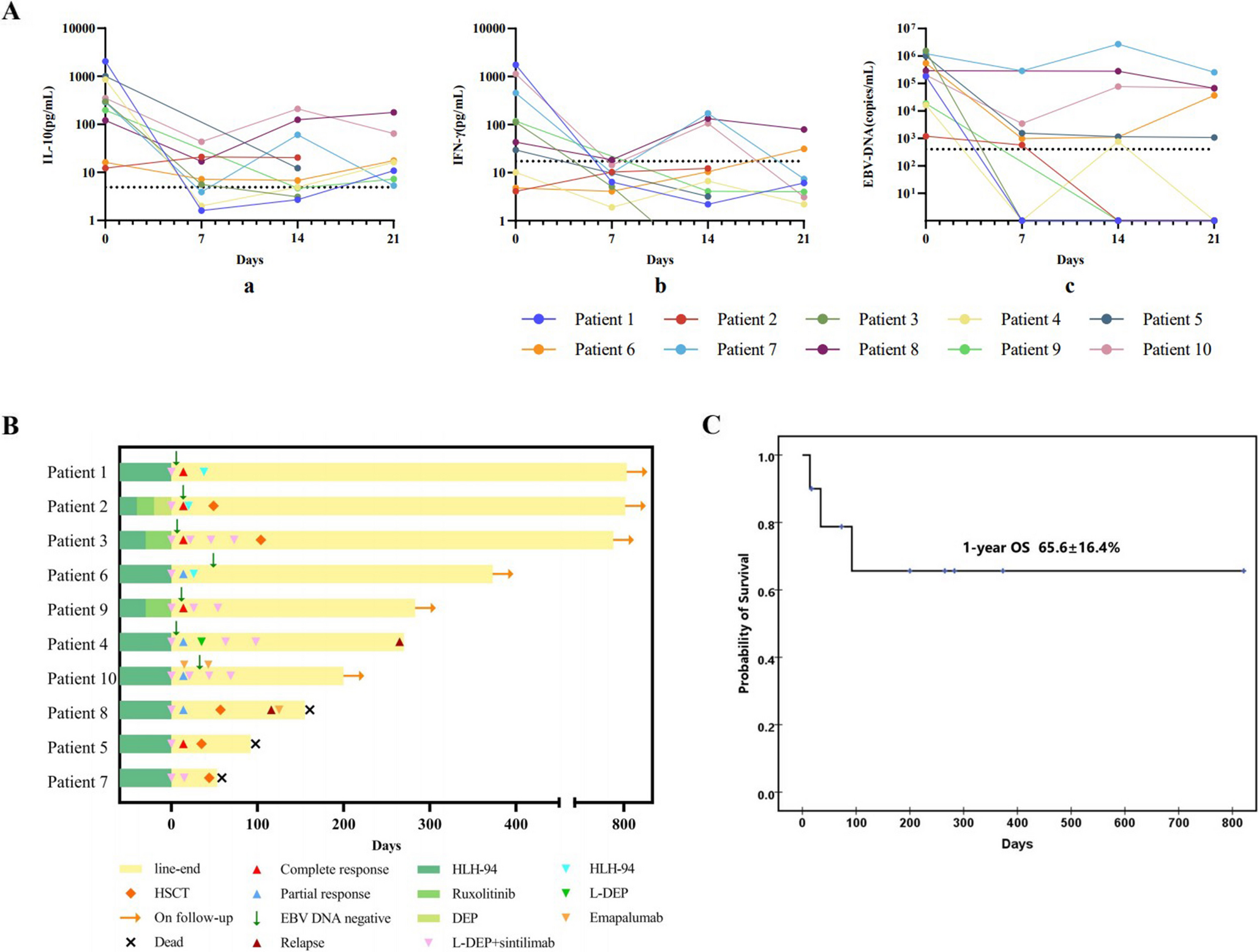
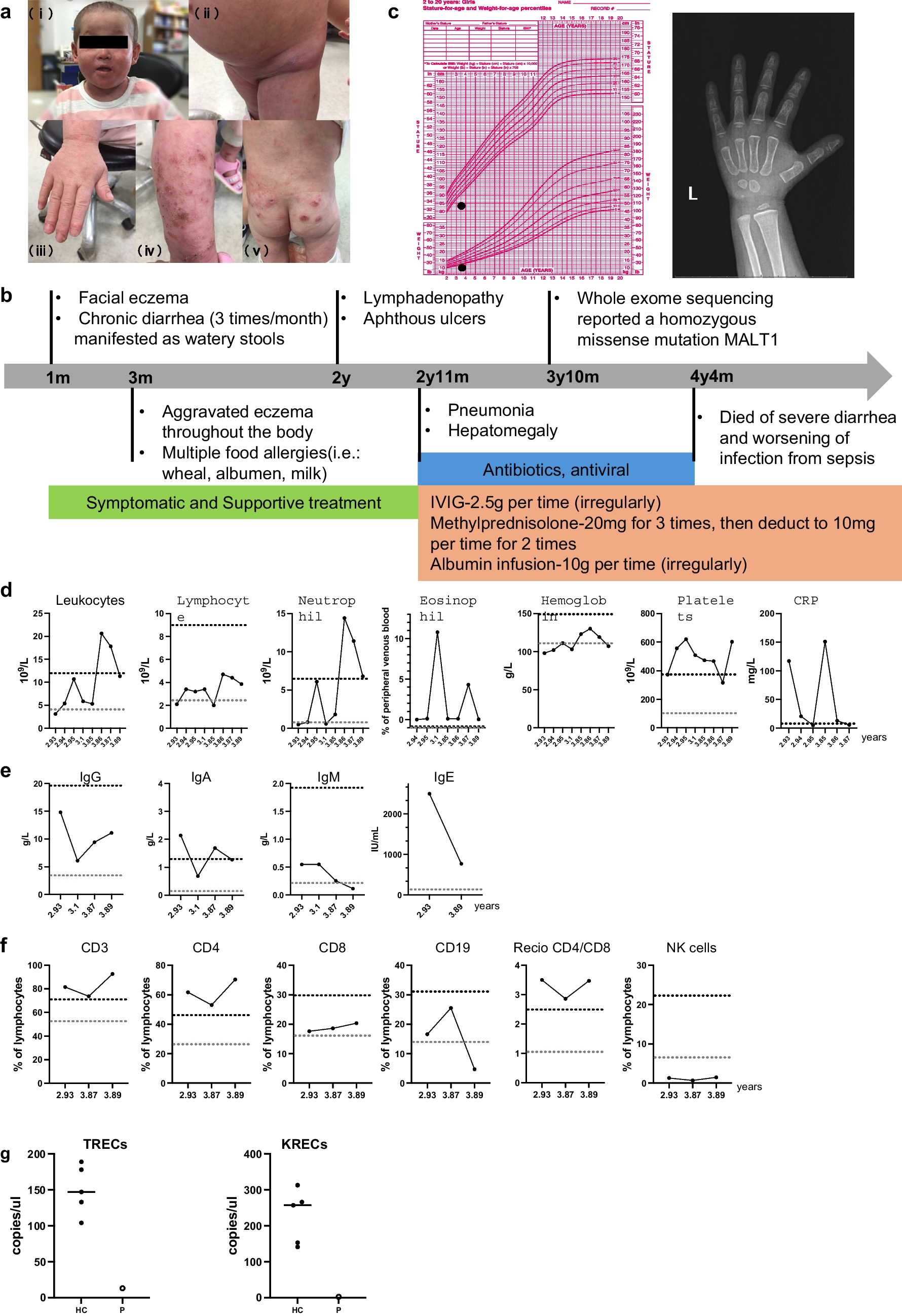
Human MALT1 deficiency is an extremely rare monogenic inborn error of immunity (IEI), classified as combined immunodeficiency [21], offering valuable insights into human immune biology. To date, only 22 cases have been described [4,5,6,7,8,9,10,11,12,13,14]. Among them, Sefer et al. reported a large cohort of MALT1 deficiency patients through a multicenter study consist of 9 patients of Kurdish ethnicity background [11]. We presented a novel case of MALT1 deficiency caused by a bi-allelic missense mutation (c.1411G > A) in the MALT1 gene, resulting in the substitution of aspartic acid with asparagine (p.D471N). This is the first case of MALT1 deficiency identified in China. Extended genetic study was conducted to confirm the homozygous mutation in this patient. Overall, the 22 reported cases of MALT1 deficiency typically presented with recurrent bacterial, viral, and fungal infections affecting the skin, respiratory system, and intestinal tract, along with aphthous ulcers, dermatitis, inflammatory bowel disease (IBD) and failure to thrive. Except for periodontal disease, our patient had all these typical symptoms. Our patient displayed persistent elevated levels of IgE and platelets, accompanying by a reduced proportion of B cells and Tregs, a severely diminished proportion of transitional B cell but normal proportions of naïve B and memory B cell subsets. Due to progressive diarrhea and septicemia, the patient passed away at the age of 4.
MALT1 plays a dual role in lymphocytes. As a scaffold protein, it associates with BCL-10 and CARD11 to form the CBM signalosome, which is essential for the activation of NF-κB transcriptional targets upon lymphocyte stimulation. The paracaspase MALT1 cleaves and removes negative regulators of NF-κB signaling, thereby promoting lymphocyte responses in NF-kB activation and in B-cell lymphoma subtypes [22]. Consistent with this, two mouse models—Malt1-/- and protease-dead Malt1PD/PD mice—have been employed to investigate the mechanisms underlying the phenotype observed in MALT1-deficient patients. Malt1-/- mice lack detectable MALT1 expression and are therefore deficient in both scaffold and protease functions. These mice exhibit agammaglobulinemia and increased susceptibility to infections yet display normal behavior and lifespan and are even protected from autoimmune inflammation [16, 22,23,24,25,26]. Our patient exhibited uniformly impaired CBM-mediated NF-κB activation in primary CD4+ and CD8+ T cells demonstrated by reduced phosphorylation of the p65 subunit, leading to deficient production of IL-2 and TNF-a. In contrast, Malt1PD/PD mice have intact MALT1 protein expression and scaffolding function but specifically lack proteolytic activity due to a knock-in mutation at the active site cysteine (C472A) [16, 22, 23, 26]. Unlike Malt1-/- mice, which typically have reduced levels of IgE and IgG antibodies, Malt1PD/PD mice develop hyper-IgE, multiorgan infiltration of lymphocytes, immune dysregulation, polyendocrinopathy, and IPEX-like gastroenteritis [16].
In line with these findings of the murine models, our patient mainly presented with IPEX-like disorders (such as progressive eczema, chronic diarrhea, consistent high IgE levels and severe food allergy) in the first two years of her life. Recurrent bacterial and viral sinopulmonary infections did not become apparent until the age of 2, and the patient ultimately succumbed to uncontrollable sepsis. Interestingly, we found a similar pattern in patients with mutations located in the caspase-like domain of the MALT1 gene [5, 7, 11,12,13]. In resemblance with previous cases, our patient exhibited severely decreased but not undetectable Treg cells. Tregs are profoundly impaired in Malt1-/- mice, with markedly reduced Treg numbers observed in the peripheral blood of young animals [17, 18, 23, 27,28,29,30,31]. However, this defect appears to be masked in aged Malt1-/- mice due to progressive peripheral Treg expansion [19, 32, 33]. Despite this compensatory expansion, aged Malt1-/- mice develop atopic dermatitis (AD)-like skin inflammation, with no evidence of inflammation in other organs [34]. Notably, hyper-IgE levels also have been reported in aged Malt1-/- mice [24]. T cells and keratinocytes were initially considered prime candidates based on the atopic dermatitis phenotypes observed in hypomorphic variants of CARD11 and CARD14. The precise cellular source contributing to this phenotype remains unclear, as conditional deletion of MALT1 in T cells (CD4-Cre) or keratinocytes (K5-Cre) did not reproduce the hyper-IgE or AD-like phenotype [35].
Another potential contributor to the inflammatory phenotype observed in MALT1-deficient patients is the inability of MALT1 to cleave HOIL1 (heme-oxidized IRP2 ubiquitin ligase 1), a negative regulator of NF-κB signaling [34]. Inactivation of paracaspase function in Malt1PD/PD mice leads to excessive interferon-γ (IFN-γ) production by effector lymphocytes, contributing to immune pathology [23]. A similar reduction of tregs was observed in Malt1PD/PD mice, where natural Treg proportions in the CD4+ T cell compartment were reduced by∼60% [16, 24]. Thus, selective inhibition of MALT1 proteolytic activity effectively skews the immune response toward autoimmunity by impairing Treg development [22, 34]. Autoimmune manifestations (including alopecia, vitiligo and psoriasis) typically in Malt1PD/PD mice were not commonly seen in MALT1 deficiency patients, occurring in only 35% of all cases. However, our patient did not show autoimmune symptoms, except she tested positive once for anti-myeloperoxidase antibody (MPO), a type of anti-neutrophil cytoplasmic antibody (ANCA) commonly associated with autoimmune diseases (data not shown).
In Malt1PD/PD murine model, there is a developmental defect in marginal zone B cells, B1 B cells, IL-10 producing B cells and mature T and B cells. In addition, Malt1PD/PD mice retained normal or partially reduced proportions of geminal center (GC) B cells, and the Tfh compartment was only partially reduced. By contrast, complete absence of GC B cells with an associated decrease in Tfh cell has been reported in Malt1-/- mice, where MALT1 has a B-cell-intrinsic role for proliferation, survival, and GC formation [16, 17, 25, 36]. In our patient, we found a normal proportion of Tfh cells, with significantly higher expression of activation markers PD-1 and ICOS. Previous studies on MALT1 deficiency from Japan indicated a significant decline in B cell numbers correlated with the age of patient, accompanied by a maturation arrest in B cells. In addition, the aberrant differentiation of B cells cannot be restored by CpG treatments [13]. To our surprise, our patient displayed normal proportion of naïve and memory B cells. In review of all the 22 reported MALT1 deficiency patients, only 7 cases had detected naïve and memory B cells [6, 8, 11, 13, 14, 20]. 2 out of 7 patients reported the percentage of naïve and memory B cells were within the normal range [11]. Furthermore, the percentage and absolute number of transitional B cell were found to be significantly reduced in our patient. Transitional B cells play an important part in regulating inflammation and tolerance [37]. Defective B cell development with a block in transitional B cell has also been announced in CARD11-deficient individuals demonstrated of imbalanced transitional B cell subsets [38]. Together, these findings highlight the role of the CBM complex in human B-cell development [37, 39, 40], indicating impaired B cell differentiation in our patient, despite the variability in B cell phenotypes observed among MALT1 deficiency patients.
Taken together, the immunological phenotypes observed in our patient support the notion that the MALT1 D471N mutation phenocopies a partial loss of both MALT1 scaffolding function and paracaspase activity. We demonstrated that the expression of both MALT1 mRNA and protein was reduced in the D471N variant. Given the variability in mRNA expression, we cannot conclude that the amino acid substitution directly causes destabilization of the MALT1 protein, as the reduced protein levels may simply reflect differences in transcript abundance. Additionally, the cycloheximide chase assay revealed that the MALT1 D471N mutant was unstable and underwent more rapid degradation compared to the WT protein. Our data suggest that the reduced expression of the MALT1 D471N variant results from both transcriptional and post-transcriptional mechanisms. In contrast, other missense mutations resulting in amino acid substitutions did not affect MALT1 gene expression, as indicated by comparable mRNA levels measured in T-cell-derived or PBMC-derived cDNA from healthy controls using RT-PCR analysis [4, 6, 7, 10]. Although many human MALT1 mutations result in either reduced (e.g., W580S, I600N, D535N, L386P, N485S, W580R) or undetectable (e.g., S89I, D184Y) MALT1 protein expression. Interestingly, MALT1 was expressed at WT levels in a homozygous variant carrying the c.2418G > C mutation, which results in the missense substitutions E795D in the MALT1A splice isoform and E806D in MALT1B. While the resulting MALT1A/B proteins lacked protease activity, MALT1A—but not MALT1B—retained its scaffolding function. This makes it the only known hypomorphic MALT1 mutation reported to date [10]. Additionally, further cases of MALT1 deficiency are needed to determine whether hypomorphic or reduced-expression mutations give rise to distinct clinical phenotypes in humans. Unfortunately, due to the limited samples and the death of our patient, MALT1 proteolytic activity could not be assessed through cleavage of known substrates such as CYLD or HOIL-1. To evaluate the impact of the D471N mutation on MALT1 protease function, future efforts to reconstitute a MALT1-knockout T cell line with the D471N mutant would be valuable to determine whether this variant can restore MALT1 proteolytic activity.
At present, severe infections remain the primary cause of poor prognosis in MALT1 deficiency. If clinically feasible, prompt hematopoietic stem cell transplantation (HSCT) is highly recommended, as it has been shown to be highly effective in rectifying immune deficiency, with successful outcomes in now 6 of 6 reported cases [5, 7, 11, 13, 20]. HSCT is therefore considered a potentially curative therapy for MALT1 deficiency.




Comments (0)