
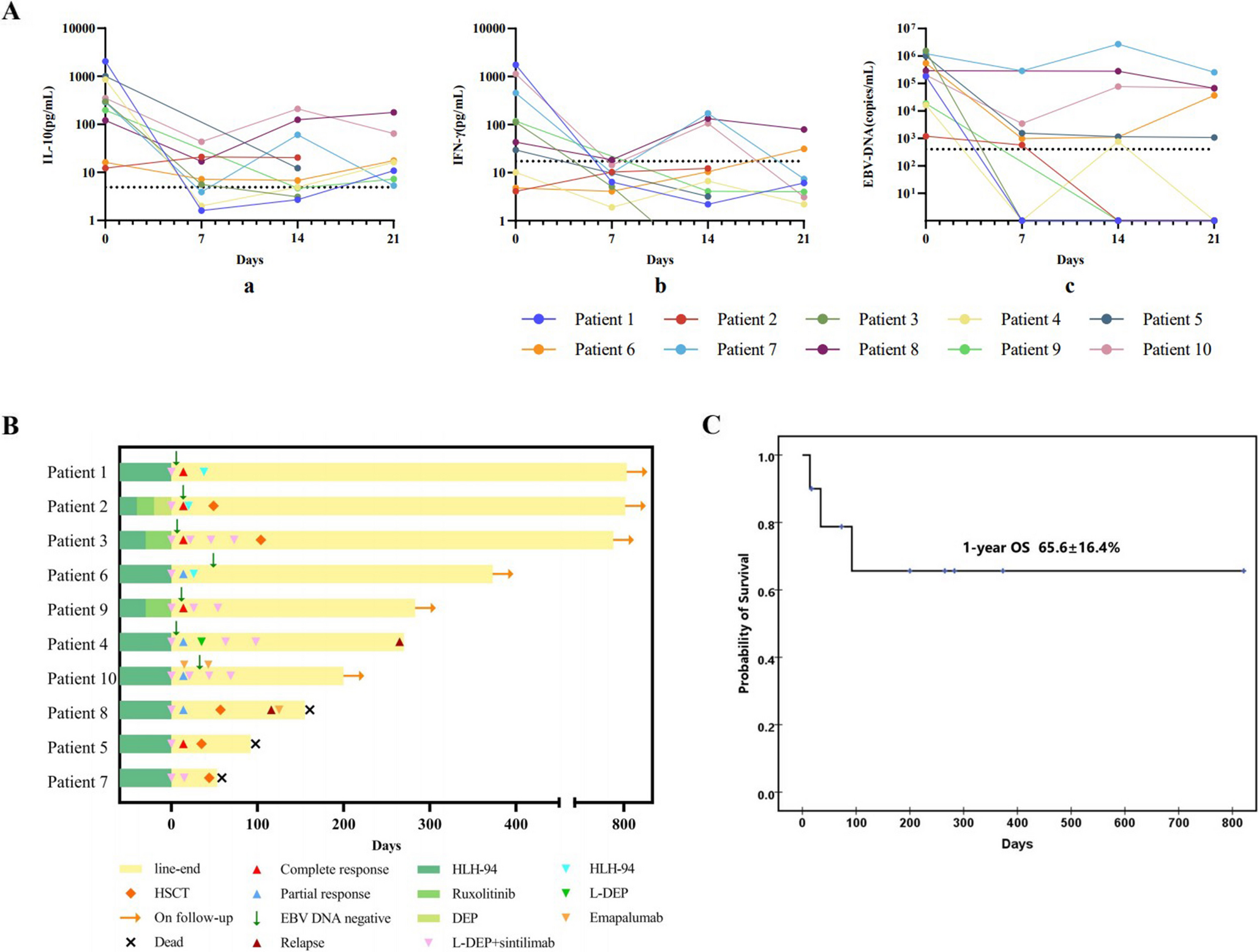
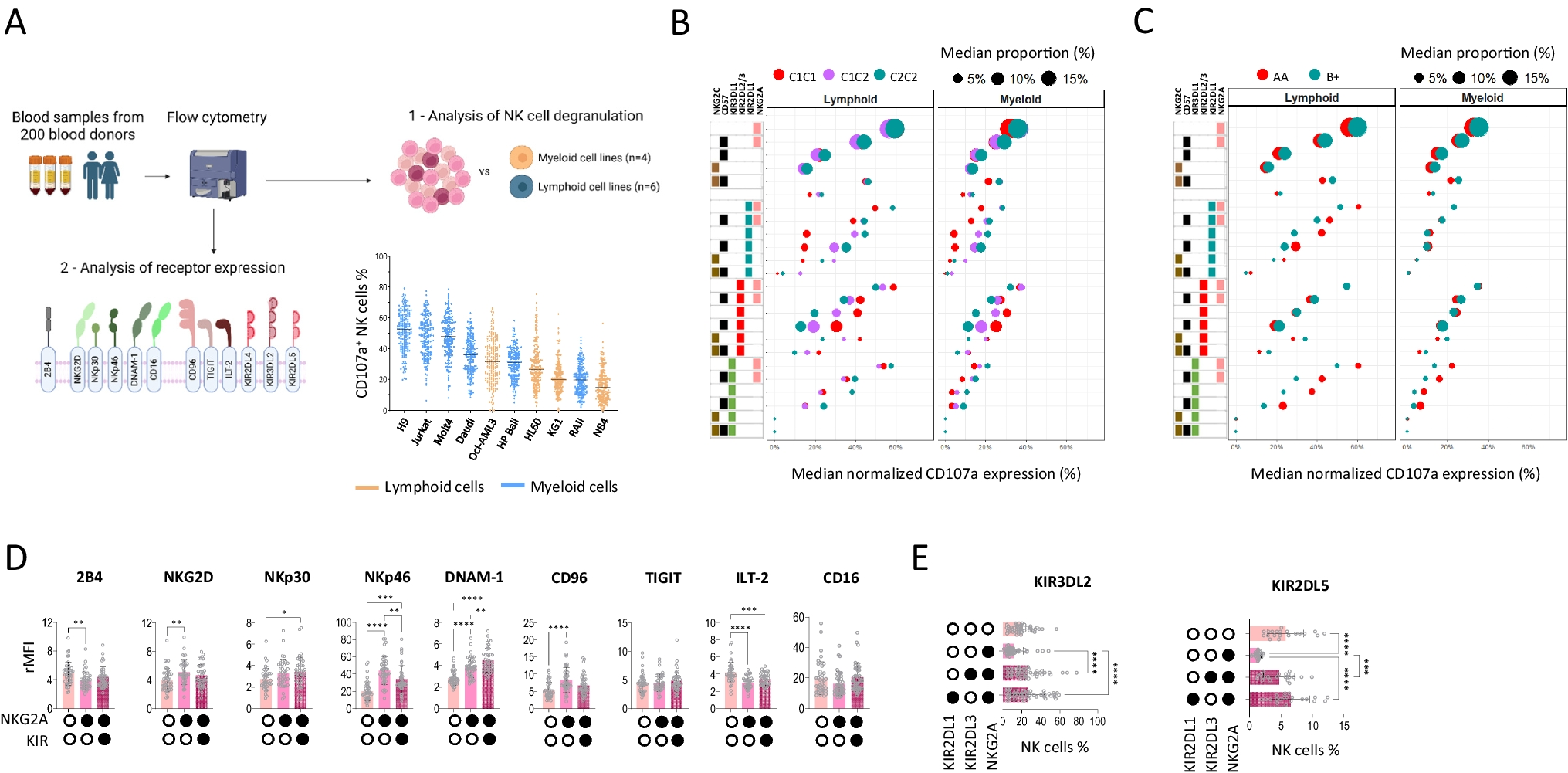
Remember me
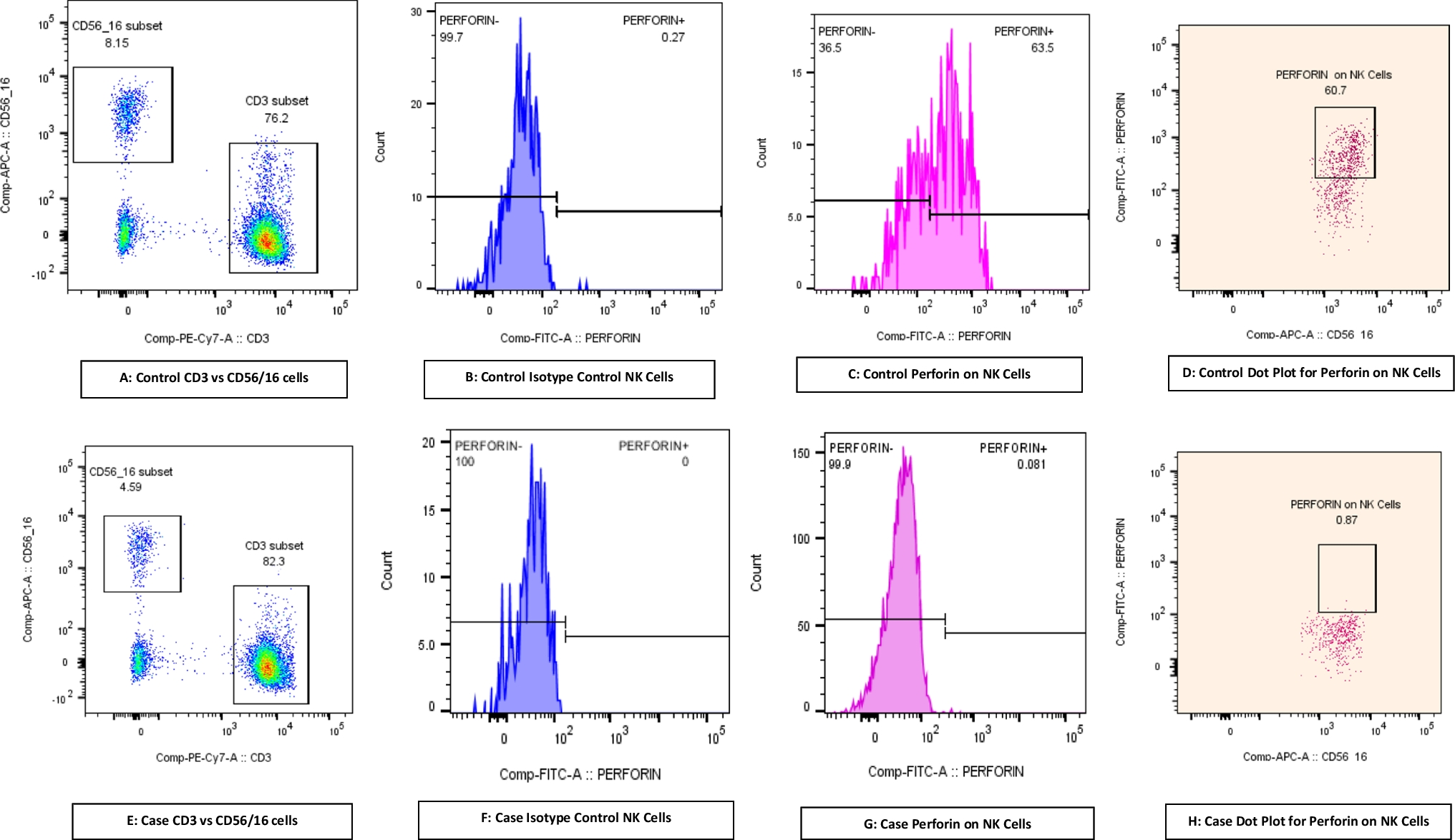


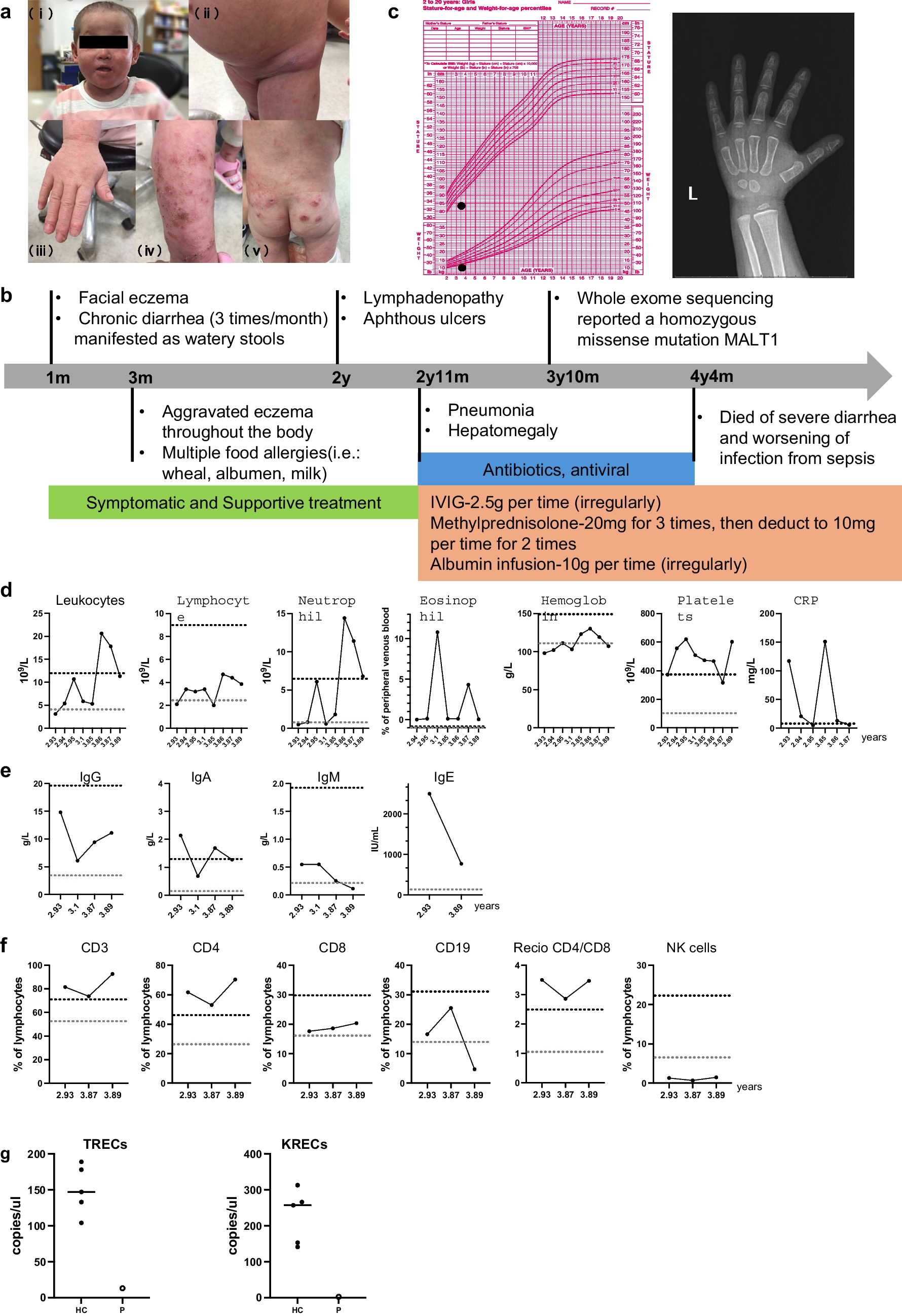

A 1-year-old girl from a consanguineous family, known for experiencing recurrent infections, was diagnosed with agammaglobulinemia following genetic testing that revealed a novel in the MALT1 gene (c.609G > A, p. Trp203). Her clinical symptoms consisted of persistent respiratory infections. Despite being treated with intravenous immunoglobulin (IVIG) therapy, her symptoms worsened. At the age of 8 months, the patient was admitted to the intensive care unit (ICU) with severe pneumonia. A computed tomography (CT) scan showed extensive pulmonary infiltration and areas of necrosis. Adenovirus was detected in the nasal swab. Immunological testing confirmed undetectable IgG levels and a complete absence of memory B cells, consistent with agammaglobulinemia. The immunoglobulin levels in the patient are as follows: IgG is at 108 mg/dl, IgA is at 5 mg/dl, and IgM is at 9 mg/dl, with a total IgE of 0.1 Ku/L. The lymphocyte subgroup percentages are CD3 + 1360, CD4 + at 900, CD8 + at 440, CD19 + at 500, and CD16+56 at 50 cells/µL. Eosinophilia and neutropenia were not detected. Six months later, she exhibited BCG scar activation (see Fig. 1), and thoracic CT revealed mediastinal lymphadenopathy. Despite negative Quantiferon and mycobacterium cultures, we initiated anti-tuberculosis therapy. Genetic analysis revealed the homozygous MALT1 mutation c.609G > A, p. Trp203, which was associated with her immunodeficiency. The genetic analysis identified a novel homozygous mutation in the MALT1 gene (c.609G > A, p. Trp203). The patients’ mutation resulted in the MALT1 protein losing its function, which hindered the activation of NF-κB and disrupted the immune response. It is a homozygous nonsense mutation that creates a premature stop codon. It has been established that similar nonsense mutations in the MALT1 gene cause premature termination of the protein and result in a loss of function. This leads to the elimination of MALT1’s protease activity, halting the activation of NF-κB signaling and causing defective cytokine production.
Fig. 1
a: The necrotizing areas of the lung, b:BCG scar activation, c:the mutation diagram
The mutation is consistent with the clinical features observed in this patient, who presented with severe, recurrent infections and agammaglobulinemia. This finding expands the known spectrum of MALT1 mutations and further emphasizes the disorder’s genetic basis. The mutation c.609G > A, p. Trp203, is a novel variant that has not been previously reported in the literature. Initial treatment involved intravenous immunoglobulin (IVIG) therapy to replace the deficient antibodies and provide temporary immune support. Despite this, the patient’s condition continued to deteriorate, and further interventions were necessary. As a result, hematopoietic stem cell transplantation (HSCT) was considered a long-term therapeutic option to restore immune function. Preparation for HSCT was initiated, which has proven an effective strategy in patients with MALT1 deficiency, offering the potential for immune reconstitution. In a study involving 19 patients with MALT1 deficiency, the clinical characteristics were in line with CID, and all individuals experienced the onset of the disease during the first six months of life. The primary clinical manifestations observed in the disease included recurrent infections (100%), skin involvement (100%), failure to thrive (FTT) (100%), oral lesions (67%), chronic diarrhea (56%), and manifestations of autoimmunity (44%). Most patients showed typical frequencies of total T and NK cells. Eight patients (89%) had mild to moderate reductions in B cell counts [5].
MALT1 deficiency is a rare and severe immunodeficiency disorder that can present with a broad spectrum of symptoms, often involving recurrent infections and immune dysregulation [4, 5]. The genetic mutation described in this case (c.609G > A, p. Trp203) is novel and extends the understanding of the genetic basis of MALT1 deficiency. While IVIG therapy can help mitigate some of the symptoms, hematopoietic stem cell transplantation (HSCT) remains the most effective treatment option for restoring immune function in MALT1-deficient patients.
MALT1 deficiency is a rare and severe form of combined immunodeficiency with significant clinical implications. Early intervention with immunoglobulin replacement therapy and consideration of HSCT is essential to improving the prognosis of affected individuals.
Comments (0)