Representativeness of the E200K gCJD case panel
Bioassays in animal models remain the gold standard for prion strain characterization, enabling the phenotyping of propagated prions based on vacuolar lesion profiles and incubation periods.
However, research focusing on the prion strain typing of E200K-associated Creutzfeldt–Jakob disease (CJD) has been fragmented and limited [7, 24, 37, 47].
In this study, we present the first comprehensive attempt to establish the prion strains responsible for E200K genetic CJD (gCJD) by analyzing 24 cases from France, Spain, and Slovakia using human PrP-expressing transgenic mice (tgMet and tgVal).
While our goal was to capture the global diversity of prion strains in E200K gCJD patients, practical limitations, such as the high cost and long duration of bioassays, and the rarity of E200K-Val129 cases, constrained the number of cases we could include. Furthermore, due to the limited availability of biological material, we were unable to bioassay different brain regions from each patient. Previous research has shown that sCJD patients can harbor multiple prion strains or strain mixtures in different brain regions, which may also apply to gCJD cases. This was not fully explored in our study due to material constraints.
Despite these limitations, our results clearly demonstrate the presence of two distinct prion strains, either alone or as mixtures, in the E200K gCJD cases. Given the number of cases (n = 22) and their geographical diversity, it is reasonable to conclude that these two strains likely represent the dominant prion variants in E200K gCJD. However, we cannot exclude the possibility that other prion strains may be implicated in some cases, particularly given the limitations in brain region sampling.
Prion strains in E200K gCJD patients
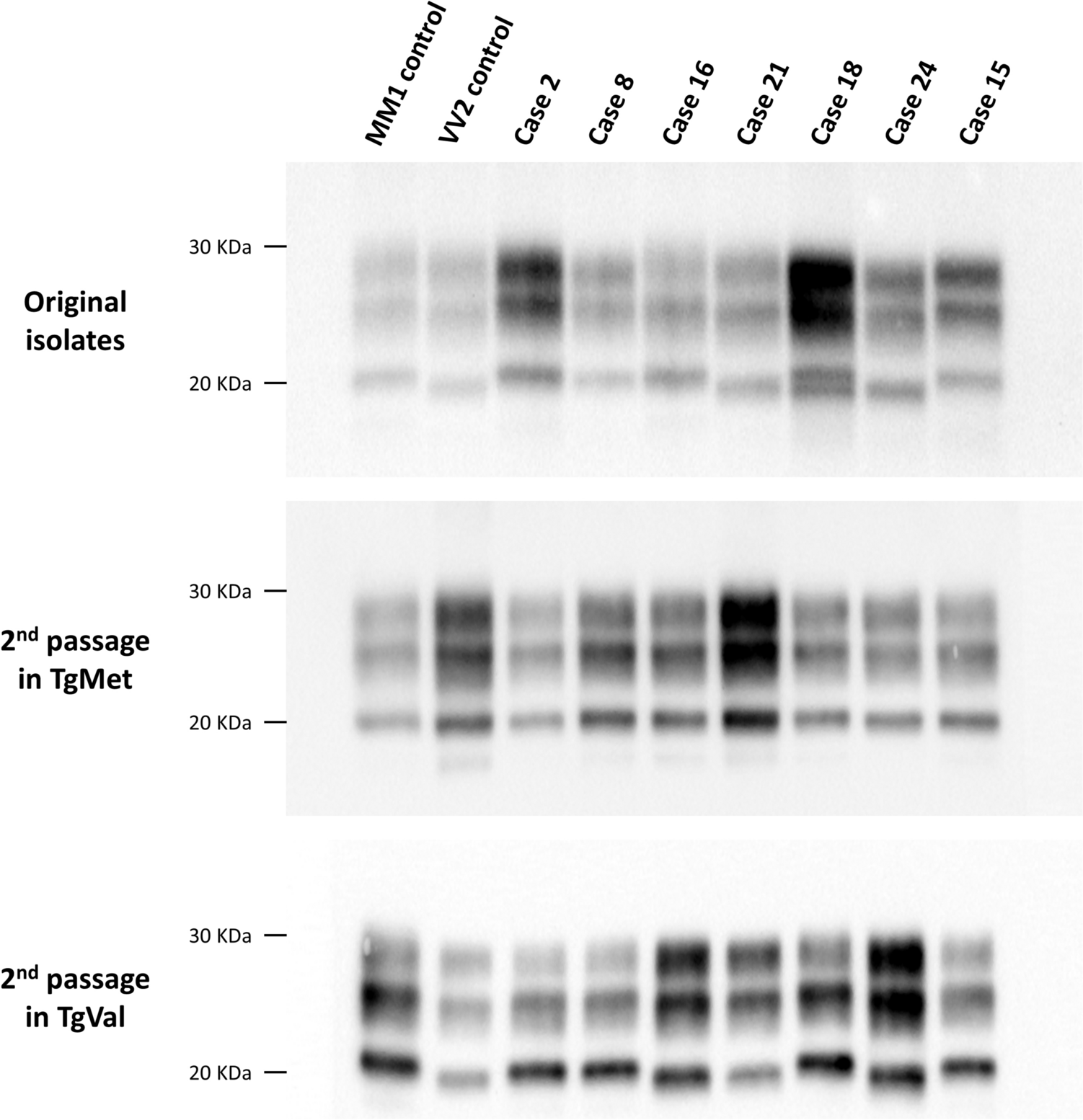
The prion strains identified in E200K gCJD brain homogenates following transmission into tgHu mouse models were identical to those previously found in sCJD patients using the same methodology [10]. Specifically, the strains correspond to M1CJD and V2CJD, which are responsible for the most common clinico-pathological forms of sCJD, predominantly observed in MM/MV1 and VV/MV2 patients, respectively [6, 10, 11, 23].
Of note, the correlation between prion strain type and PRNP codon 129 polymorphism in E200K gCJD closely mirrors that seen in sCJD. In both diseases, patients homozygous for methionine at codon 129 (Met129/Met129) harbor the M1CJD strain, while heterozygous patients (Met129/Val129) can present with either the M1CJD or V2CJD strains, or a mixture of both. In our study, this was evident in the E200K-Met129/Val129 cases, which showed a mixture of prion strains following transmission in tgHu mice.
Although we had only one case of E200K-Val129/Val129, the presence of the V2CJD strain in this case is consistent with strain-typing studies of sCJD cases homozygous for valine at codon 129, where V2CJD is typically found in Val129/Val129 patients with PrPres type 2 in the brain. While further studies are needed to confirm this observation, it suggests that the PRNP codon 129 polymorphism plays a similar role in strain selection in both gCJD and sCJD.
The apparent identity of strains between sCJD and gCJD E200K that we observed is in line with the previous transmission studies in voles showing that E200K gCJD produce disease phenotypes indistinguishable from those of sCJD type 1 [37]. It also agrees with the fact that genetic CJD forms share common neuropathological traits with sporadic cases, particularly a classification in very similar histotypes, which depend on the polymorphism at codon 129 of PRNP and the biochemical PrPres type [4], although some differences exist, mainly at the level of immunohistochemical PrP deposition patterns [27]. Whether these variations can be explained by the presence of different minor strain components in gCJD brains or by other factors remains an open and interesting question.
Role for a somatic PRNP E200K mutation in sporadic CJD
Sporadic Creutzfeldt–Jakob disease (sCJD) remains the most common form of human prion disease, but despite extensive research its underlying cause still remains unclear. It is uncertain whether sCJD originates from endogenous factors, such as the spontaneous misfolding of normal cellular prion protein (PrPC), or from exposure to an unidentified exogenous agent. Numerous case–control studies have explored various risk factors, including dietary habits, occupational exposure to animal products, and prior medical procedures. While some studies have suggested links to surgeries and blood transfusions, the evidence has been inconsistent and unable to establish definitive associations [13, 14, 35, 36, 50].
The most widely accepted hypothesis suggests that sCJD results from a random misfolding event in PrPC or a somatic mutation in the PRNP gene [44], analogous to spontaneous mutations observed in other neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS). Somatic mutations in genes linked to these diseases are increasingly identified in the brains of affected patients [29, 31, 42], suggesting a similar mechanism could underlie sCJD.
Some authors have hypothesized that the instability of the octapeptide repeat region of the PrPC protein may lead to the accumulation of somatic mutations over the lifetime, eventually initiating PrPSc formation and triggering disease [30]. Post-zygotic mutation D178N was confirmed as the cause of disease in a patient showing somatic mosaicism with negative family history [1]. Moreover, a recent study by Won et al. (2023) found a higher frequency of the E200K mutation in hippocampal and frontal cortex samples from sCJD cases compared to age- and genotype-matched controls, specifically in Met129/Met129 and Met129/Val129 variants [53]. However, contrasting evidence from McDonough et al. (2024) found no evidence of somatic PRNP mutations in neuropathologically affected brain areas from sCJD patients, as revealed by deep DNA sequencing [33].
The identification of the same dominant prion strains (M1CJD and V2CJD) in both sCJD and familial E200K gCJD patients provides an indirect argument supporting the hypothesis that somatic mutations in PRNP could be a key driver in the occurrence of sporadic Creutzfeldt-Jakob disease (sCJD). However, this hypothesis remains challenging to substantiate. Efforts to model heritable prion diseases in transgenic (Tg) mice expressing PRNP mutations that are strongly linked to genetic forms of Creutzfeldt–Jakob disease (gCJD) in humans have met with only limited success [51]. While transgenic mice expressing either the human E200K PrP variant or the mouse equivalent mutation (E199K) have been developed, these models have consistently failed to spontaneously develop an authentic prion disease [3, 7, 19, 22, 48].
Bringing definitive evidence of the role of the E200K mutation in the occurrence of sporadic Creutzfeldt–Jakob disease (sCJD) will therefore require further extensive investigation. This will likely include advanced techniques, such as single-cell sequencing to detect low-frequency somatic mutations, longitudinal studies of brain tissue, and more sophisticated animal models that better replicate the spontaneous development of prion diseases.
E200K as a proxy to study preclinical sCJD
Our findings, which reveal a clear overlap in prion strains between E200K gCJD and sCJD, along with comparable prion seeding activity and infectivity patterns in peripheral tissues, strongly indicate that these two conditions share significant pathological similarities. This positions E200K gCJD as a highly informative model for studying the early stages of sCJD pathogenesis, potentially offering critical insights that could enhance both the understanding and clinical management of sporadic CJD cases.
Given the rare incidence of sCJD in the general population, it is challenging to conduct prospective studies on individuals prior to disease onset. However, the high penetrance of the E200K mutation allows for longitudinal studies in carriers, offering an opportunity to explore the preclinical phase of disease development. This could facilitate the identification of early biomarkers and the development of diagnostic tests that may eventually benefit both at-risk individuals and the broader population.
The age at disease onset and clinical progression in sCJD are strongly influenced by disease subtype [41]. Our finding that prion strains found in E200K gCJD cases align with disease subtypes in a manner mirroring patterns seen in sCJD could further aid in refining prognostic tools for patients, ensuring a more personalized approach to disease management.
Additionally, animal model studies have clearly demonstrated that the efficacy of anti-prion treatments is heavily influenced by both the timing of therapeutic intervention and the specific prion strain involved. In this context, conducting therapeutic trials in E200K mutation carriers, who are at higher genetic risk, could not only provide direct benefits to these individuals but also offer crucial data for optimizing treatment strategies for sCJD patients. By understanding the timing and strain-specific effects of potential therapies, these trials could inform the best practices for treatment in sporadic cases as well.
The observed similarities in prion strains between E200K gCJD and sCJD emphasize the need for continued research into strain diversity and its value in the understanding of prion diseases. Further studies involving PRNP mutation carriers with a high risk of developing CJD could offer essential guidance for developing targeted therapies and improving care for those affected by these devastating diseases.





Comments (0)