
Remember me
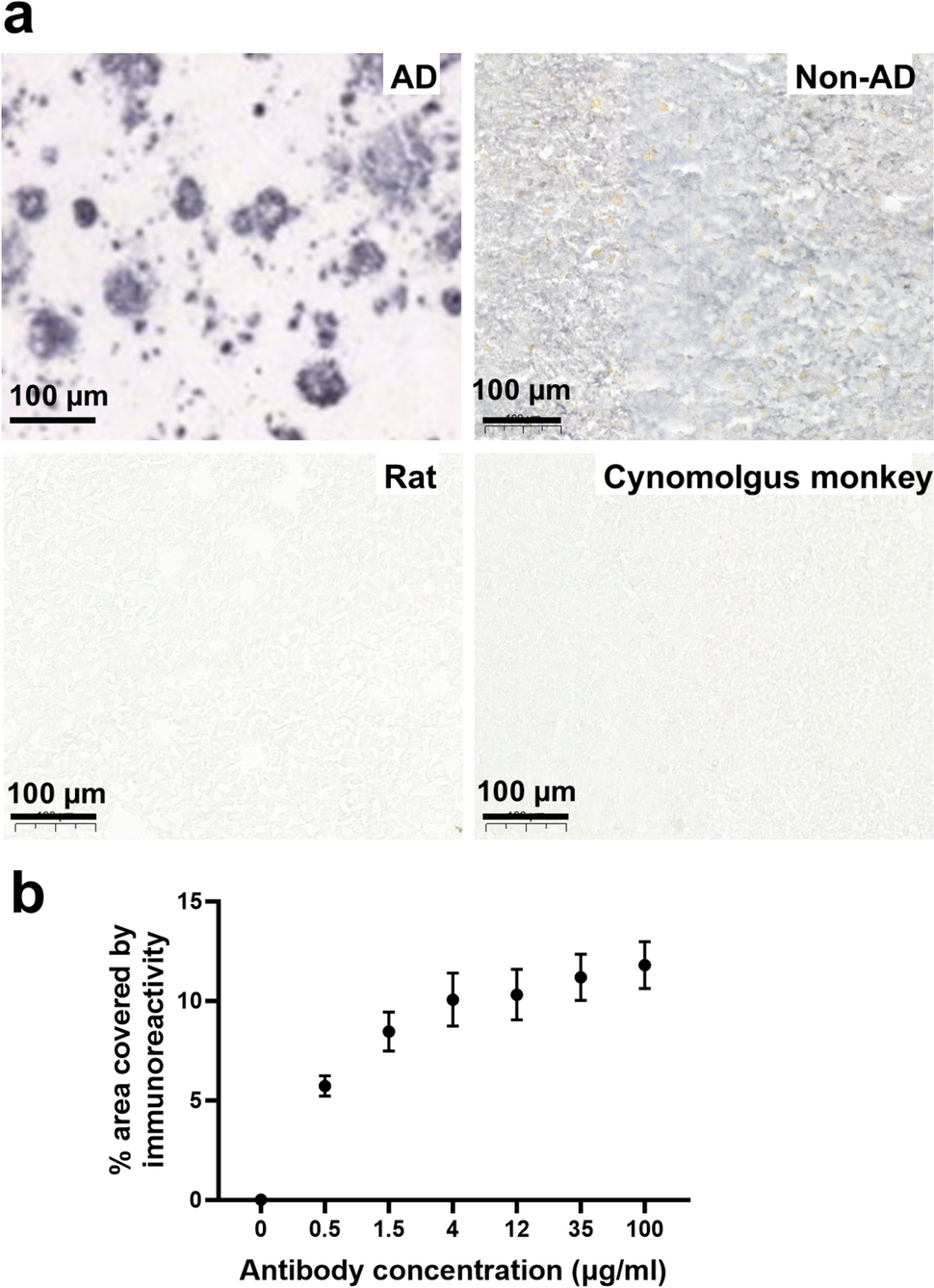




Key resources are listed in Supplementary Table 1.
Study designTwo miRNA-based short-hairpins (miR5 and miR7) targeting distinct regions of rat Atp10b mRNA (regions targeted on Atp10b mRNA miR5: 5’ TCCTGGTGATTCTGAACTGGAT 3’, miR7: 5’ TCCTAAGACAGTGCCTATATAT 3’) were packaged in an adeno-associated vector (AAV2/7) under a neuronal promoter (CMVenhanced-Synapsin) and unilaterally injected in the right SNpc of adult female Wistar rats. We have previously reported that these two miRNA-based short-hairpins efficiently KD ATP10B in mouse cortical neurons cultures [16]. A vector with a scrambled sequence (SCR: 5’ AATACGACGGTAAGTGAGTACG 3’) that has no equivalent target in the Rattus norvegicus genome was used as control. The AAV2/7 viral vectors were produced by the Leuven Viral Vector Core [36]. The long-term study consisted of 15–16 rats per treatment group. Following stereotaxic surgery, behavioral evaluation and in vivo striatal dopamine transporter (DAT) binding using 18F-FE-PE2I and positron emission tomography imaging (PET) were performed. Rats were euthanized 1 year post-injection to investigate neuropathological changes in the brain or changes in protein expression. A separate cohort consisted of 6 rats per treatment group and were sacrificed after 1 month without behavioral evaluation to examine possible short-term alterations by immunohistochemistry. Additionally, a smaller group of rats (2–4 per treatment group) were euthanized after 2 weeks to assess the efficiency of vector transduction in dopaminergic neurons. An overview of the study design is shown in Fig. 1.
Fig. 1
Targeted knockdown of ATP10B in adult rat SNpc using AAV vectors. miRNA-based short-hairpins (miR5 and miR7) targeting distinct regions of Atp10b mRNA, and a scrambled non-targeting sequence (SCR) packaged in AAV2/7 vector under a neuronal promoter (CMVenhanced-synapsin) were stereotactically injected in the right SNpc of rats. a Overview of the protocol followed in this study and vector constructs. b Schematic representation of the Atp10b mRNA sequence, with the full mRNA sequence shown in green, coding sequence (CDS) in red, and exons indicated in black. The binding sites for miR5 and miR7 are highlighted in blue and purple, respectively. c, d The percentage of double TH + RFP + cells within the total TH + population in the SNpc was determined for the three vectors 2 weeks post-injection. c Representative image of the SNpc of a rat injected with SCR, miR5 and miR7 vector 2 weeks post-injection, fluorescently stained with TH and RFP antibodies, shows efficient transduction of dopaminergic neurons. d Each data point represents an individual animal, and the number of TH + and TH + RFP + cells from 7 sections was quantified using stereological methods
AnimalsAdult (8–9 weeks old) female Wistar rats weighing 200–250 g (Janvier, France) were used for this study, and housed (2–3 animals per cage) in individually ventilated cages with free access to food and water, under a normal 12 h-light/12 h-dark cycle. Animal experiments were carried out in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC) and approved by the Bioethical Committee of the KU Leuven (Belgium) (ECD project 161/2022). As a limitation, findings reported in this manuscript relate to female rats, as male rats were not included in this study. Similar experiments in male rats would be important to generalize our findings to both sexes.
Stereotaxic surgeryAll surgical procedures were performed using aseptic techniques. Rats were anaesthetized with ketamine (60 mg/kg, intraperitoneal (i.p.), Nimatek, Dechra, Belgium) and medetomidine (0.4 mg/kg, i.p., Domitor, Orion Pharma, Finland), and placed in a stereotactic frame (Stoelting, Wood Dale, IL). Rats were injected unilaterally with 3 µL of AAV2/7-CMVenhsynapsin-mCherry-miRSCR, AAV2/7-CMVenhsynapsin-mCherry-miR5, or AAV2/7-CMVenhsynapsin-mCherry-miR7 in the right SNpc (AP: – 5.3 mm; L: – 2.0 mm; DV: – 7.2 mm calculated from dura, using bregma as reference), using a 30-gauge needle and a 10 μl Hamilton syringe (Hamilton, Bonaduz, GR, Switzerland). Each vector was injected at a normalized titer of 7.5 × 1011 GC/mL (2.25 × 109 GCs injected per animal), at a flow rate of 0.25 μL/min. Following injection, the syringe was left in place for an additional 5 min, and then slowly retracted.
Behavioral testsDifferent behavioral tests were performed to assess motor asymmetry and motor function. For each test, rats were acclimatized to the testing room at least 1 h prior to assessment. Automated systems were used for the rotarod and open-field tests. Quantification of the videos for the other tests was done by a blinded researcher.
Rotarod testMotor coordination and balance was assessed using an accelerated rotarod system (IITC Life Science Rotarod Model I-755, Campden Instruments). Before surgery, rats were trained for 5 min at a constant speed of 5 rpm. During this initial training phase, rats were placed back on the rod after falling. In the second phase of training, rats underwent 3 trials of 1 min at a fixed speed of 5 rpm, 10 rpm, and 15 rpm, with 5 min of rest in-between trials. For testing rotarod performance at baseline, and post-surgery, rats were positioned on rotating rods with a progressively increasing rotation speed, ranging from 4 to 40 rpm over a 5 min period. Three trials were conducted at every timepoint with 5 min resting interval in between. The rotarod protocol was performed on three consecutive days, with the average of the three days taken for statistical analyses.
Open field testThe open field set-up consisted of a square box (1 m × 1 m) surrounded by opaque walls that prevent observation of visual cues outside the arena. Rats were placed in the center and spontaneous behavior was recorded using an overhead camera for 5 min. Total distance traveled, velocity, and % immobility time were obtained using AnimalTracker [9], and ipsi-/contralateral 180° turning and rearing frequency were calculated manually in a blinded manner on the acquired videos.
Cylinder testThe cylinder test was employed to quantify asymmetry in forelimb use. Contacts made by each forepaw with the wall of a 20-cm-wide clear glass cylinder were scored from the videotapes by an observer. A minimum of 20 contacts were recorded and quantified for each animal. The number of contralateral forelimb contacts was expressed as a percentage of total forelimb contacts. Rats not performing enough contacts were excluded from the analysis.
Elevated body swing testIn the elevated body swing test (EBST), the rat tail was taken approximately 3 cm from its base, and the animal was elevated to about 5 cm above the cage floor and held along the vertical axis for 5 s. Ipsilateral or contralateral swing were counted manually in a blinded manner, whenever the animal moved its head out of the vertical axis to either side by at least 30° within the 5 s observation interval. The EBST comprised five trials of maximum 5 s, each followed by a brief rest. The mean for all five trials was used for statistical analysis.
Catalepsy testThe catalepsy bar test is used to measure the failure to correct an imposed posture resulting from muscular rigidity. For this test, the forepaws of the rats were placed on an elevated bar with the hind paws remaining on the floor. The time for the rat to correct this posture was recorded and used an index of the intensity of catalepsy. Three trials were performed, with a brief rest in between. The average of all three trials was used for statistical analysis.
Small animal DAT microPET imagingFor longitudinal in vivo measurement of DAT integrity, we used 18F-FE-PE2I, a highly selective DAT PET radioligand [37]. 18F-FE-PE2I produced under GMP was obtained from the hospital radiopharmacy at UZ Leuven.
Rats were anesthetized (5% isoflurane in O2 for induction and 2% thereafter at 1 L/min flow rate), placed on a heated mat and cannulated with a 23G catheter. They were then placed on the imaging bed (Molecubes, Ghent, Belgium) with integral temperature and respiration monitoring before being transferred to a β-cube microPET scanner (Molecubes/Bruker, Ghent, Belgium). Dynamic PET images were acquired for 90 min starting from intravenous injection with 18F-FE-PE2I (8.9 ± 2.7 MBq)/rat. Specific activity was 286 ± 119 GBq/micromol with mass doses injected between 0.1 and 0.4 nmol. Rats were kept under anesthesia during the entire procedure (2.5% isoflurane in O2 at 1 L/min flow rate), with temperature and respiration monitored throughout. After PET scanning, a computed tomography (CT) image was acquired for anatomic coregistration with an X-cube CT scanner (Molecubes, Ghent, Belgium), using the following parameters: 50kVp, 480 exposures, 85 ms/projection, 100 μA tube current, rotation time 60 s. After scanning, rats were recovered, with on average two weeks between scanning sessions.
Image processing and analysisPET list mode data were binned into 16 frames (4 × 15 s, 4 × 1 min; /1 × 5 min /5 × 10 min and 2 × 15 min) and reconstructed into a 192 × 192 image matrix with 0.4 mm voxel size, using 30 iterations using the native Maximum-Likelihood Expectation–Maximization (MLEM) algorithm. Correction was done for randoms, scatter, attenuation based on CT, and data were scaled to kBq/cc after calibration to a standard 18F-phantom. Images were decay-corrected to the start of the scan. CT data were reconstructed using a regularized iterative algorithm [34] with a voxel size of 200 µm (isotropic) and were scaled to Hounsfield Units (HUs) after calibration against a standard air/water phantom. PET and CT data were then cropped to the skull and CT scans were co-registered to an in-house CT skull template using an affine transformation. The first 9 frames of PET data were summed for rigid co-registration with the CT scan. Dynamic PET data were then transformed to template space using the CT-based transformation matrices. Striatal and cerebellar regions of interest in template space [39] and time-activity curves were created (operations carried out using PFUS (https://store.bruker.com/products/pfus-image-fusion-remote/)). Time activity curves were imported in PKIN, where a standard reference tissue model (SRTM) was applied to generate non-displaceable binding potential (BPnd) values as measure for absolute tracer binding, using the cerebellum as the reference tissue [41]. Parametric BPnd images were then generated using the voxel SRTM method in PXMOD (PMOD Technologies GmbH, Zürich, Switzerland) with the same reference region.
RNAscope in situ hybridizationFor the staining of the brain (in situ hybridization and immunostaining), rats were sacrificed with an overdose of sodium pentobarbital (200 mg/kg, i.p., Dolethal, Vetoquinol, Lure, France), and transcardially perfused with saline and 4% paraformaldehyde (PFA) in PBS. After post-fixation overnight in 4% PFA, 20 µm thick coronal brain sections were made with a vibrating microtome (HM 650 V, Microm).
For RNAscope in situ hybridization, brain sections were transferred to a 48-well plate and washed once with PBS containing 0.1% Tween-20. Sections were then incubated with RNAscope™ Hydrogen Peroxide (Advanced Cell Diagnostics) for 10 min at room temperature (RT), protected from light. Tissue sections were mounted onto Superfrost™ Plus slides (Thermo Fisher Scientific), and were allowed to air-dry for 1 h at RT, then baked for 30 min at 60 °C to ensure proper adhesion of the tissue. Sections were then post-fixed for 1 h at 4 °C in 4% PFA in PBS. After fixation, sections were dehydrated using an ethanol gradient (50%, 70%, 100%, and 100%, 5 min each). The day after, target retrieval was performed using a steamer and 1X Target Retrieval Reagent solution (Advanced Cell Diagnostics) for 10 min. Next, protease digestion was performed using RNAscope Protease III reagent (Advanced Cell Diagnostics, V2 assay) for 30 min at 40 °C. For RNA detection, probe targeting Atp10b RNA from Rattus norvegicus (RNAscope Probe—Rn-Atp10b, catalog # 1155561-C1, Advanced Cell Diagnostics) was added to the brain sections. An additional section was incubated with RNAScope 3-plex Negative Control Probe (RNAscope 3-plex Negative Control Probe, catalog # 320871, Advanced Cell Diagnostics) which binds to DapB, a gene present in Bacillus subtilis strain. For signal amplification, RNAScope Multiplex FL v2 amplifiers (Advanced Cell Diagnostics) were added. Following probe hybridization and amplification steps, fluorescent signal detection was performed using the RNAscope Multiplex Fluorescent v2 Assay (Advanced Cell Diagnostics). For channel 1 (C1) development, RNAscope Multiplex FL v2 HRP-C1 reagent was applied to each section and incubated at 40 °C for 15 min. Slides were then washed twice in 1X RNAscope Wash Buffer for 2 min each at RT with gentle agitation. Tyramide signal amplification (TSA) was performed using TSA Plus Cyanine 5 (TSA 650, PerkinElmer), diluted 1:1500 in 1X RNAscope Multiplex TSA Diluent. Slides were incubated at 40 °C for 30 min. Following fluorophore development, slides were washed twice in 1X Wash Buffer (2 min each). To quench residual horseradish peroxidase (HRP) activity and prevent cross-reactivity in subsequent detection channels, RNAscope Multiplex FL v2 HRP Blocker was applied to each section and incubated for 15 min at 40 °C. Slides were then washed twice with 1X Wash Buffer for 2 min each at RT. RNAScope assay was combined with TH and RFP fluorescent staining. After the RNAscope protocol, we proceeded with fluorescent staining as explained in ‘Immunohistochemical staining’ Sect. below.
AnalysisRNAscope analysis was performed on either TH + or RFP + cells using ImageJ. Regions of interest (ROIs) were manually delineated by carefully outlining individual TH + or RFP + cells with clearly distinguishable nuclei that did not overlap with neighboring cells. The “Find Maxima” tool in ImageJ (Prominence > 10) was used to automatically detect and quantify Atp10b RNA puncta within each ROI. For the analysis of RFP + cells, the total number of Atp10b puncta detected in all the ROIs of one section was divided by the number of ROIs, resulting in an average puncta count per cell in one SN section.
Immunohistochemical stainingImmunohistochemistry (chromogen) staining and immunofluorescence staining were performed on free-floating sections. Sections were washed with PBS, and incubated in an antigen retrieval solution (0.1 M citrate buffer pH 6.0) for 30 min at 80 °C. After 20 min on ice, the sections were washed in PBS, and incubated in 3% H2O2 and 10% methanol in PBS for 10 min at RT. The sections were washed 2 × 5 min in PBS with 0.1% Tergitol (PBS-T), and blocked with 10% goat serum, or 10% donkey serum depending on the species of the secondary antibodies used, for 1 h at RT. Then sections were incubated overnight in primary antibody diluted in PBS-T with serum at RT. The next day, the sections were washed 2 × 5 min in PBS-T, followed by biotinylated secondary antibody for 1 h at RT (for immunohistochemistry chromogen staining) or fluorescent secondary antibody for 2 h at RT (for immunofluorescence staining). Then, for immunohistochemistry chromogen staining, the sections were washed 2 × 5 min in PBS-T and incubated with streptavidin–horseradish peroxidase complex. Following 2 × 5 min washes in PBS-T, TH immunoreactivity was visualized using DAB 3,3’diaminobenzidine tetrahydrochloride. For immunofluorescence staining, after being rinsed in PBS and allowed to dry, the sections were coverslipped with Mowiol.
TH Immunofluorescence of the SNpc for stereological quantifications was performed on sections mounted on Superfrost plus glass slides, dried overnight and following an antibody signal enhancement (ASE) protocol [25]. Sections were pretreated with antigen retrieval solution (Tris–HCl-EDTA buffer pH 9.0 + 0.05% SDS) in the steamer for 30 min. After 20 min on ice, sections were washed with ASE wash buffer (PBS + 0.5% Tween-20) and blocked for 30 min with ASE blocking solution (PBS + 2% donkey serum, 50 mM glycine, 0.05% Tween-20, 0.1% Tergitol, 0.1% BSA). Sections were incubated overnight at 4 °C with primary antibody in ASE primary antibody buffer (PBS + 10 mM glycine, 0.05% Tween-20, 0.1% Tergitol, 0.1% H2O2). Next day, after one rinse and 2 × 3 min washes with ASE wash buffer, sections were incubated for 2 h at RT with secondary antibody diluted in ASE secondary buffer (PBS + 0.1% Tween-20). After being rinsed in PBS and allowed to dry, the sections were covered with Mowiol.
See Supplementary Table 1 for antibody information.
AnalysisImages of dSTR were captured with the Aperio CS2 slide scanner. QuPath software was employed to measure TH Optical Density (OD) in 6 sections per animal across the dSTR (sampling frequency of every twelfth sections, in a rostro-caudal manner).
The number of TH + /RFP + cells at 2 weeks post-injection (Fig. 1) and the number of TH + cells in the SNpc at 1 month and 1 year post-injection (Fig. 5) was determined by stereological measurements using the optical fractionator method in a computerized system as described before [2], in a blinded manner. The software used was Stereo Investigator (MicroBrightField, Delft, The Netherlands), and the hardware Leica Application Suite 3.6. The parameters were the following: frame area = 20 000 μm2, frame height = 10 μm, guard height = 2 μm, fame spacing = 300 μm, thickness = 17–19 μm, coefficient error = 0.09–0.17. For each animal, we analyzed 7 sections throughout the entire SNpc (sampling frequency of every tenth sections, in a rostro-caudal manner).
The number of TH + cells and HuC/D + cells in the SNpc (Fig. 6) was determined by automatic cell detection using QuPath software. For each animal, we analyzed 3–4 sections at different levels of the SN. Brain sections were imaged on a Zeiss Axioscan Z.1 slidescanner equipped with a Zeiss Colibri 7 illumination source and a Hamamatsu Orca Flash 4.0 V3 camera. Images were taken with a 10 × Plan-Apocromat objective (NA 0.45), at a sampling rate of 0.650 μm/pixel. The setup was controlled by ZEN blue (software version 3.8, Carl Zeiss Microscopy GmbH).
The number and volume of the late endosomes/lysosomes was determined in TH + dopaminergic neurons (Fig. 7) in 3 sections per animal spanning the SNpc. Images were captured using a Nikon AX confocal microscope at 60 × magnification. The captured Z-stacks were 3D reconstructed using Imaris version 10.0.1 (Oxford Instruments, Abingdon, United Kingdom), and labeled organelles were quantified using Imaris Spot detection within delineated TH + cell surfaces within the field of view.
Protein extraction and Western blotFor protein expression analysis, rats were sacrificed with an overdose of sodium pentobarbital (200 mg/kg, i.p.), and transcardially perfused with saline. SN tissue was freshly isolated, snap-frozen and stored at – 80 °C until analysis. Samples were weighed and homogenized in 10 volumes of RIPA buffer (50 mM Tris–HCl, 150 mM NaCl, 0.1% (w/v) SDS, 1% (v/v) Triton-X100, 0.5% (w/v) Sodium Deoxycholate, 1.0 mM EDTA pH 7.4) containing a protease inhibitor cocktail (Roche complete) and phospho-STOP EASYPACK (Roche) using a tissue homogenizer (TH, Omni Tissue Homogenizer). After homogenization, samples were sonicated 3 times during 15 s, and centrifuged at 6000 g for 10 min at 4 °C. Protein sample concentration was determined by BCA protein assay (Thermo Scientific, MA, USA) according to the manufacturer’s directions.
Western blots were performed using 15 µg of SN extracts prepared in 4 × Laemmli buffer (0.24 M Tris pH 6.8, 7.27% SDS, 40% Glycerol, 10% β-Mercaptoethanol, 0.01% Bromophenol blue). Protein samples were loaded on 4–15% Criterion™ Tris–HCl Protein Gel and transferred to a polyvinylidene fluoride membrane (Bio-Rad). Nonspecific binding sites were blocked for 1 h at RT in 5% nonfat milk in PBS-T. After overnight incubation at 4 °C with primary antibodies, blots were washed 3 × 10 min with PBS-T and incubated with horseradish peroxidase-conjugated secondary antibody for 1 h. After 3 × 10 min with PBS-T, bands were visualised using Clarity Western ECL (Bio-Rad) and developed with a GE ImageQuant 800 (GE Healthcare). Densitometric analysis was performed using ImageQuant.
See Supplementary Table 1 for antibody information.
Generation of isogenic knockout linesThe human induced pluripotent stem cells (iPSCs) BJ SiPS-D TH-TdTomato line was cultured using StemflexTM medium (ThermoFisher). Before nucleofection, the cells were pre-treated with a 10 μM Rhok inhibitor for an hour. The cells were then dissociated using Accutase, after which they were pelleted and resuspended in 800 μl PBS containing 5 μg of px330 CRISPR DNA each. Finally, they were transferred into nucleofector cuvettes. Nucleofection was performed using the P4 Nucleofector kit from Amaxa and the standard and program hiPSC CA-137. Two CRISPR sgRNA targeting exon1 were used to produce the knockout (KO) (CR1: CACCGCTACAACTTGACACAGCAG, AAACCTGCTGTGTCAAGTTGTAGC CR2: CACCGAATTGCTCAAAGAGATTCCG, AAACCGGAATCTCTTTGAGCAATTC), genotype primers used were F: TGGCAGTGGAGAGTCAGAGA, R: CCTGGGGAACAGAATGAGAC. Clones with homozygous or compound heterozygous deletions, leading to truncations and frameshift mutations, were identified using genotyping PCR. Clones for all lines containing deletions were identified by Sanger sequencing. Two generated ATP10B KO clones were used in this study, termed ATP10B KO clone#1 and ATP10B KO clone#2.
Midbrain differentiationHuman iPSCs were cultured in StemflexTM medium (ThermoFisher) at 37 °C, with 5% CO2 in a humidified incubator, as previously described. Differentiation was done by dual-SMAD inhibition with SB431542 (R&D Systems, 10 μM), LDN193189 (Stemgent, 100 nM), B27 minus Vit A and N2 in DMEM-F12. Midbrain-specific patterning for midbrain NPCs was made with the addition of CHIR99021 (Stemgent, 3 μM), Purmorphamine (STEMCELL, 2 μM), and SAG (Abcam, 1 μM). Post patterning Neural maturation medium was DMEM F12 medium containing N2, B27-VitA, 20 ng/mL GDNF (R&D Systems), 20 ng/mL BDNF (R&D Systems), 0.2 mM ascorbic acid (Sigma), 0.1 mM dibutyryl cAMP (Biolong), 10 μM DAPT (Cayman Chemical). The medium for long-term culture was DMEM F12 medium containing N2, B27-VitA, 10 ng/ML GDNF (R&D Systems), 10 ng/mL BDNF (R&D Systems), 0.2 mM ascorbic acid (Sigma).
Staining and quantificationCells were fixed with 4% PFA for 20 min, blocked in 0.1% Triton X-100 in 5% horse serum/PBS, and then incubated in primary antibodies overnight at 4 °C. The following day, cells were washed and incubated in secondary antibodies and DAPI nuclear stain according to protocol. Imaging was performed using the High-content imager CX7 with phenotyping and quantifications using CellProfiler. The average of at least three fields of a well was counted as the N value for statistical analysis.
See Supplementary Table 1 for antibody information.
Time-course FACS analysisMidbrain neuronal cultures or iPSC cultures were dissociated at different time points with Accutase, resuspended in PBS supplemented with 1xB27, DNase, and 10 μM Y-27632 ROCK inhibitor, and filtered through a 35 μm strainer into a 5-ml Falcon round-bottom tube. The BD Celesta analyzer (BD Biosciences) was used to measure TH-TdTomato-positive cells. The flow cytometry results were analyzed using FlowJo™ v10.8 Software (BD Life Sciences).
Live-cell caspase-3/7 activity assayActive caspase-3/7 activity was detected using the LIVE/DEAD™ Image-iT™ Caspase-3/7 Detection Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, iPSC-derived neuronal cultures were incubated with the caspase-3/7 detection reagent at 37 °C for 1 h to label cells undergoing apoptosis. Following live staining, cells were immediately fixed with 4% paraformaldehyde for 15 min at room temperature. Fixed cells were then subjected to immunofluorescence staining.
Statistical analysisData are presented as mean ± standard error of the mean (s.e.m.), and individual values. Statistical analyses were performed using GraphPad Prism version 10.2.0. For analyzing paired observations within one group of animals, we used a parametric paired two-tailed t-test or non-parametric Wilcoxon signed-rank test. For all the other analyses, we employed either a one-way ANOVA, or two-way ANOVA, followed by post-hoc comparisons. Correlation analyses were performed using Pearson correlation. Parametric or non-parametric statistical tests were performed based on the normality of residuals and equal variance across groups, tested using the D’Agostino-Pearson omnibus and Brown-Forsythe tests respectively. The α-value was set at 0.05.
Comments (0)