
Remember me
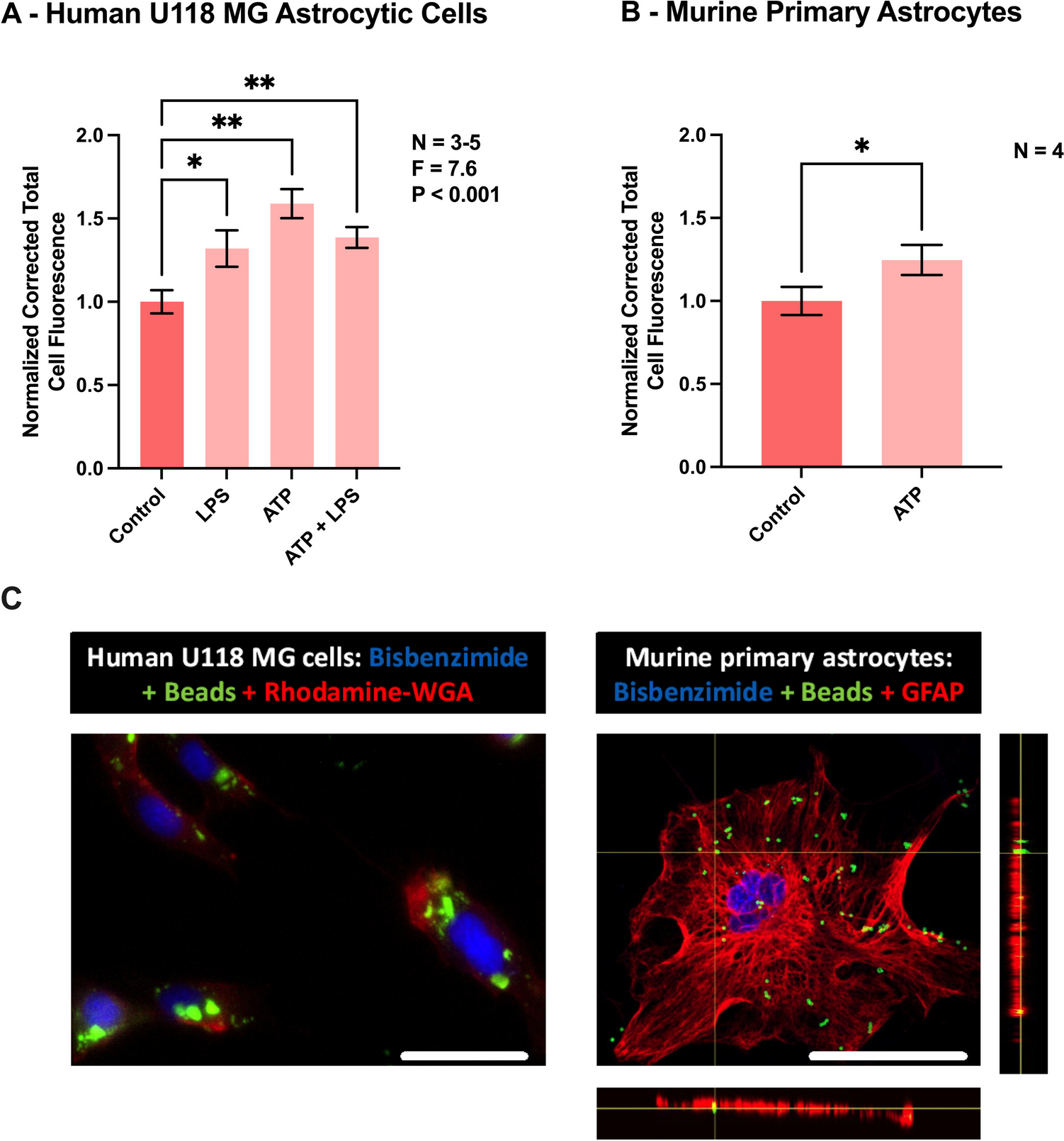
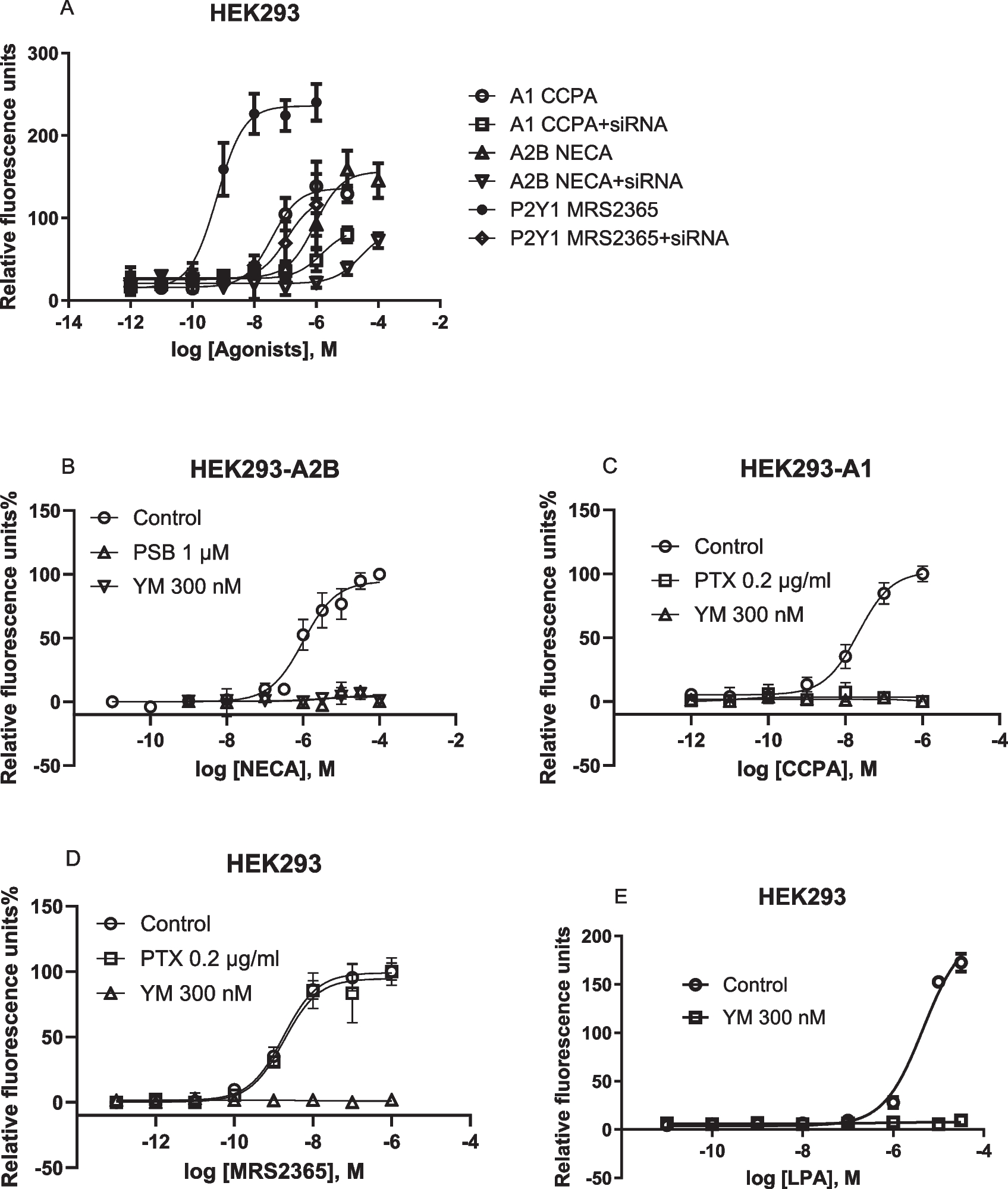
Here, we further explore the Gαi-Gβγ-PLCβ-Ca2+ signaling axis, previously demonstrated to be inhibited by Gαq/11 inhibitor FR and Gq/11 knockout [16, 18]. In addition to FR, Gq/11 siRNA and another Gq/11 inhibitor, YM, inhibited calcium mobilization in HEK293 cells, endogenously expressing the Gq-coupled P2Y1R, or overexpressing the recombinant human Gi-coupled A1 or Gi- and Gs-coupled A2B ARs (Fig. 1). These findings are consistent with previous pharmacological results [15] and align with the proposed mechanism that Giβγ-mediated calcium signaling is under Gq/11 control in HEK293 cells [16]. Thus, the results from the present study using FR, YM, Gq/11 siRNA knockdown and prior CRISPR-Cas9-based Gq/11 knockout research [16] are all consistent.
Fig. 1
Gq/11 siRNA knockdown and effects of Gq/11 inhibitors in HEK293 cells. A. Effects of Gq/11 siRNA (250 nM) on A2B-, A1-, and P2Y1 receptor-mediated intracellular calcium mobilization in HEK293 cells. Data are mean ± SEM from three independent experiments. B,C,D,E. The recombinant A1 and A2BARs are stably expressed in HEK293 cells. The P2Y1 and LPA receptors are endogenously expressed in HEK293 cells. CCPA, A1 agonist; NECA, A2BAR agonist; MRS2365, P2Y1R agonist. Data expressed as mean ± SEM from 2–4 independent experiments
Unlike the Gq inhibitors FR and YM, treatment with the Gi inhibitor PTX had no effect on Gq/11-coupled P2Y1R agonist MRS2365-induced Ca2+ release in HEK293 cells, but completely inhibited the A1AR agonist CCPA-induced Ca2+ signaling in HEK293-A1 cells (Fig. 1C,D). The A2BAR-mediated increase of ERK1/2 activity in HEK293-A2B cells has been reported to be Gi-dependent by Yang et al. [26] based on PTX sensitivity, which was later confirmed with both PTX and CRISPR-Cas9-based Gq/11 knockout [18]. However, A2BAR-mediated Ca2+ mobilization in HEK293-A2B cells is Gi-independent, as incubation with 200 ng/ml PTX for 18 h did not affect the NECA-induced Ca2+ increase [19].
In HEK293 cells, there is a possibility that the overexpression of Gi-coupled A1 or Gs-coupled A2B ARs enable their stronger coupling to Gαq/11, and thus Gαq/11 can subsequently control the A1AR-mediated Giβγ-Ca2+ signaling. FR inhibition of native Gq/11-coupled P2Y1R-induced calcium release is expected. In addition, LPA (lysophosphatidic acid)-induced intracellular calcium mobilization (EC50 = 4.59 µM) was also completely inhibited by 300 nM YM (Fig. 1E).
HEK293 cells: Similar potencies of Gαq/11 inhibitors on Giβγ- versus Gαq-mediated stimulation of intracellular calcium mobilizationThe A1AR and P2Y1R are Gi-coupled and Gq-coupled receptors, respectively. We compared the potencies of FR and YM in inhibiting A1AR (overexpressed)-triggered calcium mobilization by A1AR agonist CCPA (1 µM) and native P2Y1R-mediated Ca2+ increase by P2Y1R agonist MRS2365 (1 µM) in HEK293 cells (Fig. 2A,B). The IC50 values FR and YM to inhibit P2Y1R-mediated calcium mobilization were 13.2 ± 3.2 and 12.8 ± 4.1 nM, respectively. The IC50 values for the inhibition of the A1AR-mediated effect were 8.66 ± 3.23 and 12.9 ± 2.52 nM, respectively. Additionally, the IC50 values of YM in inhibiting the effects of 10 µM carbachol and 1 µM CCPA in CHO cells overexpressing the recombinant Gi-coupled M2 muscarinic or A1 receptor, respectively, were determined to be 9.53 and 11.6 nM, respectively (Fig. 2C,D). Thus, the potencies of FR and YM for inhibition of Gq- and Giβγ-mediated calcium signaling are similar, and the use of either inhibitor at 100–300 nM should completely inhibit both Giβγ- and Gαq-triggered Ca2+ signaling.
Fig. 2
Actions of FR and YM at Gq-coupled P2Y1 and Gi coupled A1 and M2 receptors. Potencies of FR and YM in inhibition of Gαq- (HEK293 a native P2Y1R) or Giβγ-mediated calcium increase. A. MRS2365 (1 µM)-induced Ca2+ in HEK293 cells expressing a native P2Y1R (Gq coupled). B. CCPA (1 µM)-induced Ca2+ increase in HEK293 cells expressing the recombinant A1AR (Gi-coupled). C. CCPA (1 µM)-induced Ca2+ increase in CHO cells expressing the recombinant A1AR. D. Carbachol (10 µM)-induced Ca2+ increase in CHO cells expressing the recombinant M2 muscarinic receptors. Data are from at three independent experiments. The IC50 values (nM) are listed in the text
HEK293 cells: Gαq/11 inhibitors lack effects on Gi-mediated inhibition or Gs-mediated stimulation of cAMP accumulationAs previously demonstrated in other cell types [15], FR and/or YM did not affect A1AR-mediated inhibition or A2AAR- and A2BAR-mediated stimulation of cAMP accumulation in HEK293 cells (Fig. 3), although they inhibited Ca2+ signaling induced by those receptors.
Fig. 3
Effects of FR and/or YM on Gi-mediated inhibition or Gs-mediated stimulation of cAMP accumulation in HEK293 cells. Data are from three separate experiments. A. Inhibition by A1 agonist CCPA of forskolin-stimulated cAMP accumulation. B. NECA-induced cAMP accumulation. C. A2A agonist CGS21680-induced cAMP accumulation
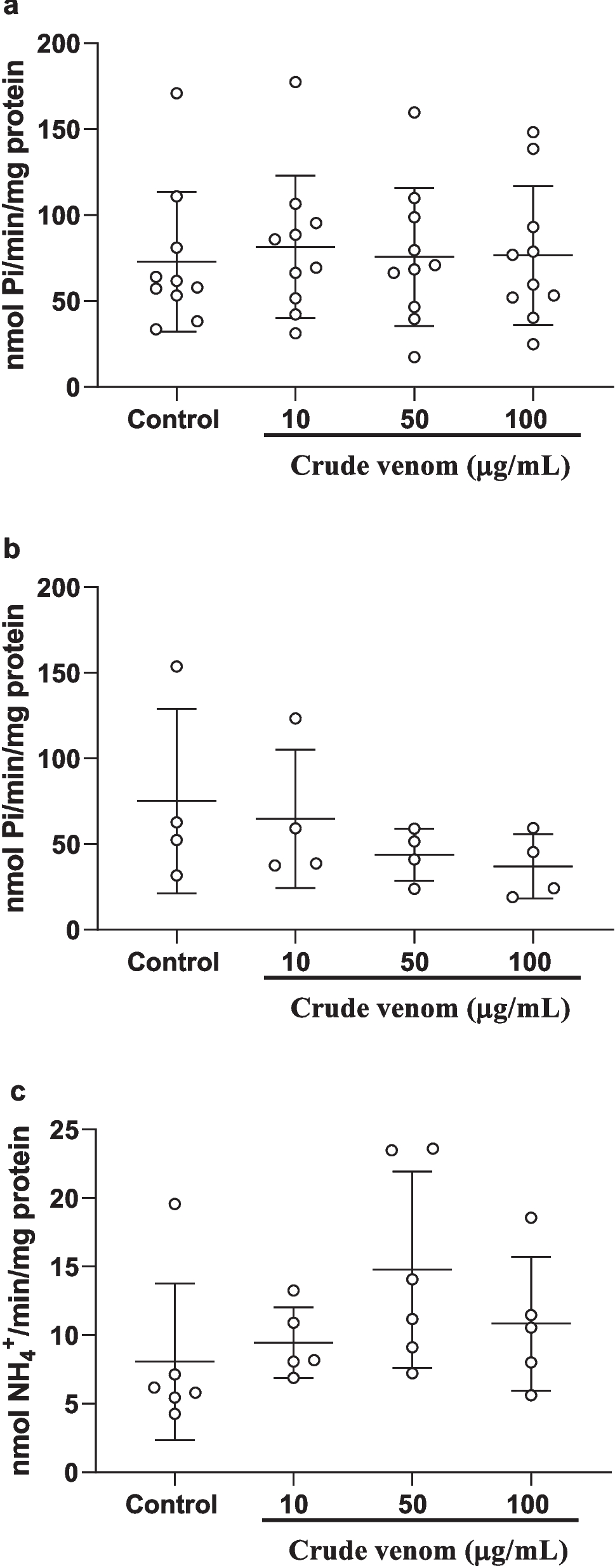
T24 bladder cancer cells: Gq/11 proteins do not play a major role in native A2BAR-mediated intracellular calcium mobilizationAs described above, the Gq/11 pharmacological inhibitors and siRNA diminished the A2BAR-mediated calcium signaling in HEK293 cells overexpressing the recombinant hA2BAR, which is consistent with results from CRISPR-Cas9-based Gq/11 knockout HEK293 cells [9]. We next carefully examined the intracellular calcium mobilization triggered by the native A2BAR in T24 bladder cancer cells [8, 22]. Figure 4A shows that, unlike in HEK293 cells, Gq/11 siRNA suppressed P2Y1R agonist MRS2365-induced calcium mobilization but not A2BAR agonist NECA-triggered calcium mobilization (Fig. 4B). The EC50s of NECA in the absence and presence of Gq/11 siRNA were 234 ± 61 and 247 ± 46 nM, respectively (n = 3), which are not significantly different (P > 0.05, Student’s t-test).
Fig. 4
Roles of Gq/11 proteins in intracellular calcium mobilization in endogenously expressed GPCRs in T24 cells. A,B. Gq/11 siRNA on P2Y1R agonist MRS2365- and A2BAR agonist NECA-induced a Ca2+ increase mediated by native P2Y1 and A2B receptors, respectively. C,D. The effect of Gq/11 chemical inhibitor YM (C) FR (D). Results are from 2–4 independent experiments
Similar to the effect of Gq/11 siRNA, Fig. 4C shows that the Gq/11 inhibitor YM (300 nM) also completely inhibited the effect of P2Y1R agonist MRS2365 but had little if any effect on A2BAR-induced calcium mobilization in T24 cells. The EC50s of NECA in the absence and presence of YM were 427 ± 76 and 524 ± 88 nM, respectively (n = 3), which are not significantly different (P > 0.05, Student’s t-test). Figure 4D shows that, similarly to YM in T24 cells, another Gq/11 inhibitor FR (300 nM) also completely inhibited calcium mobilization triggered by the native P2Y1R but not by the native A2BAR.
T24 bladder cancer cells: Contribution of both Gi and Gs proteins to intracellular calcium mobilization triggered by native A2BARsFigure 5A shows that, unlike YM and FR, PTX diminished A2BAR agonist NECA-mediated calcium mobilization in T24 cells but had no effect on the P2Y1R agonist MRS2365. Thus, it is suggested that Gi plays a prominent role in A2BAR-mediated intracellular calcium mobilization in T24 bladder cancer cells. Considering that Giα isoforms do not play a significant role in activation PLCβ [27], it is assumed that Gβγ is responsible for this effect [15]. Interestingly, simultaneous inactivation of Gi and Gs by PTX and CTX almost completely eliminated A2BAR-triggered Ca2+ release (Fig. 5A), suggesting both Giβγ and Gs are responsible for A2BAR-mediated Ca2+ mobilization in T24 cells. The EC50s of MRS2365 in the absence and presence of PTX were 1.38 ± 0.26 and 1.55 ± 0.47 nM, respectively, which are not significantly different (P > 0.05, Student’s t-test). The EC50s of NECA for Control group, PTX group and PTX + CTX group were 538 ± 112, 3550 ± 890 and 12,300 ± 4600 nM, respectively, which are significantly different (P < 0.05, One-Way Analysis of Variance followed by Bonferroni’s multiple comparison tests). The maximal effect of NECA (in terms of relative fluorescence units) in Control group, PTX group and PTX + CTX group were 505 ± 78, 173 ± 39 and 88 ± 15, respectively, which are significantly different (P < 0.05, One-Way Analysis of Variance followed by Bonferroni’s multiple comparison tests).
Fig. 5
Roles of Gi and Gs proteins in intracellular calcium mobilization mediated by endogenous GPCRs in T24 cells. Data are from three independent experiments. A. Effects of Gi inactivator PTX (200 ng/ml) and the combination of PTX and Gs inactivator CTX (500 ng/ml). B. effects of Gs inactivator CTX (500 ng/ml), EPAC inhibitor ESI09 (10 µM), or PKA inhibitor H89 (10 µM)
To examine whether the pathway downstream of Gs, PKA or EPAC, contributes to A2B-Gs-mediated calcium mobilization in T24 cells, PKA inhibitor H89 and EPAC inhibitor ESI09 were used. Figure 5B shows that both EPAC and PKA are involved in A2BAR-Gs-mediated calcium mobilization. However, the combination of ESI09 and H89 did not produce an effect larger that ESI09 alone (P > 0.05, Student’s t-test). The inactivation of Gs by CTX produced a similar effect to that of ESI09.
In addition to the A2BAR, T24 cells also endogenously express the β2-adrenergic receptor. Figure 6 shows that the β2 adrenergic agonist formoterol and isoproterenol induced a robust calcium response albeit a little less efficacious compared to the A2BAR agonist NECA. However, unlike the effects of FR on the A2BAR agonist NECA-triggered Ca2+ release, FR (300 nM) diminished Ca2+ increase triggered by both β2-adrenergic receptor agonists formoterol and isoproterenol, suggesting a role of Gq/11 (Fig. 6A,B).
Fig. 6
Effects of Gαq inhibitors FR and YM, Gi inactivator PTX and Gs inactivator CTX on endogenous GPCR-mediated Ca2+ increase in T24 cells. A,B. FR (300 nM) on triggered Ca2+ increase triggered by β-adrenergic receptor agonists isoproterenol and formoterol in T24 cells. C. Effects of YM (1 µM), CTX (500 ng/ml) and PTX (200 ng/ml). Data are from three independent experiments
In a separate set of experiments, YM (1 µM) was shown to inhibit 78% of the maximum effect (Emax) of formoterol (Fig. 6C). Unlike its effect on the A2BAR, PTX treatment did not produce significant attenuation of the Emax in β2 agonist formoterol-induced calcium mobilization (Fig. 6c). However, CTX treatment produced an effect larger than it was on the NECA-induced Ca2+ response (Fig. 6C; Fig. 5B) in the same cell type, i.e. T24 bladder cancer cells. Thus, even in the same cell type, the involvement of G proteins in intracellular calcium mobilization is dependent on receptor type, although both the A2BAR and the β2-adrenergic receptor have been reported to couple to both Gs and Gi [9, 28]. We were not able to observe formoterol-triggered Ca2+ increase in HEK293 cells.
In addition to T24 cell, the A2BAR has been reported to trigger intracellular calcium mobilization in a human breast cancer cell line, MDA-MB-231 [23], although the G proteins involved have not been examined. In the present study, we compared the effect of CTX, PTX and YM. Figure 7 shows that, unlike in T24 bladder cancer cells, YM, CTX, and PTX all inhibited NECA-induced Ca2+ increase although to a different extent. Thus, Gi, Gs and Gq are all involved in the A2BAR-mediated Ca2+ increase in MDA-MB-231 cells. Furthermore, CTX and PTX together produced an effect larger either alone (Fig. 7B). The selective A2BAR antagonist PSB603 (1 µM) completely blocked the effect of NECA (Fig. 7B). A2A-selective agonist CGS21680 stimulated cAMP accumulation in MDA-MB-231 cells (EC50 2240 ± 850 nM), but less potently than NECA with EC50 73.1 ± 18.2 nM (Fig. 7C).
Fig. 7
Effects on agonist-triggered Ca2+ increase in MDA-MB-231 breast cells expressing endogenous adenosine receptors. A. Effects of YM (300 nM), CTX (500 ng/ml) and PTX (200 ng/ml) on the response to NECA. B. Effects of A2B antagonist PSB603 (1 µM) and the combination of CTX and PTX on NECA-triggered Ca2+ increase. C. Comparison of cAMP accumulation induced by A2A agonist CGS21680 and A2B agonist NECA. D. Gene expression level of four ARs on MDA-MB-231 cells. Results are from three experiments performed in duplicate
Thus, we demonstrated that intracellular calcium mobilization in two cell types, i.e. T24 bladder cancer and MDA-MB-231 breast cancer, both expressing the native A2BARs, is independent and dependent on Gq/11, respectively. A cAMP accumulation assay confirmed that the A2BAR rather than A2AAR is the dominant AR subtype (Fig. 7C). The gene expression levels of four AR subtypes in MDA-MB-231 cells are shown in Fig. 7D with A2BAR being the highest expressed.
T24 bladder cancer cells: Involvement of PKC in A2BAR-mediated intracellular calcium mobilizationWe have demonstrated previously in HEK293-A2B cells that the activation of PKC by PMA completely diminished the Ca2+ mobilization induced by the A2BAR agonist NECA [10]. Figure 8 shows that in addition to Gs, Giβγ, and Gαq/11, PKC also plays a role in receptor-triggered cytosolic calcium increase. The PKC activator PMA completely eliminated P2Y1R-mediated calcium mobilization, but only partially diminished A2BAR-mediated calcium mobilization in T24 cells (Fig. 8A). Unlike the effect of PKC on the A2BAR, but similar to its effect on the P2Y1R, cell treatment with PKC activator PMA (1 µM) completely diminished the intracellular calcium mobilization induced by β2 agonists formoterol (Fig. 8B) and isoproterenol (Fig. 8C). Thus, the effect of PKC on calcium intracellular mobilization in T24 cells is receptor type-dependent. Pretreatment of cells with the PKC inhibitor GO for 20 min eliminated the effect of PMA at both A2BAR and P2Y1R (Fig. 8D). Interestingly, the PKC inhibitor GO alone significantly enhanced the Emax of calcium mobilization induced by P2Y1R agonist MRS2365 (P < 0.05, One-Way ANOVA) but not by the A2BAR agonist NECA (Fig. 8E,F), which suggests that PKC could act as an endogenous suppressor of P2Y1R-calcium signaling in T24 bladder cancer cells.
Fig. 8
Role of protein kinase C in A2BAR-, P2Y1- and β2-adrenergic receptor-triggered Ca2+ increase in T24 cells. A. Effects of PKC activator PMA on MRS2365- or NECA-triggered Ca2+ increase. B. Effects of PMA or PKC blocker GO (Go6983). C. Effects of PMA. D. Effects of preincubation with GO for 20 min followed by addition of PMA for 20 min before agonist addition. E,F. Effects of PKC inhibitor on P2Y1- or A2B-triggered Ca2+ increase. Results are from three experiments. #Significantly different from control (P < 0.05)
T24 bladder cancer cells: β-Arrestins 1 and 2 are not involved in A2BAR-mediated intracellular calcium mobilizationWe have shown previously that β-arrestin2 siRNA diminished P2Y1R agonist MRS2365-induced and β-arrestin2-mediated ERK1/2 activity [29]. Here it is shown that neither β-arrestin1 nor β-arrestin2 siRNA affected A2BAR agonist NECA-induced Ca2+ increase (Fig. 9).
Fig. 9
Effects of β-arrestin siRNA on A2BAR agonist NECA-triggered Ca2+ increase in T24 cells. Data are expressed as mean ± SEM from three experiments. Results are from three experiments. The transfection of siRNA was performed using Lipofectamine 2000
T24 bladder cancer cells: L-type calcium channels are not involved in A2BAR-mediated intracellular calcium mobilizationFigure 10 shows that the potency of the A2BAR agonist NECA-triggered Ca2+ increase was reduced about only twofold by the calcium blocker nifedipine (10 µM) but completely inhibited by the PLC inhibitor U73122 or the A2BAR antagonist PSB603, which supports a mechanism of intracellular mobilization.
Fig. 10
Effects of the L-type calcium channel blocker nifedipine, A2B antagonist PSB603 and the PLC inhibitor U73122 on A2B agonist NECA-triggered Ca2+ increase in T24 cells. Results are from three experiments. Inhibitors were incubated with cells for 20 min in 96-well black plates before addition of NECA
Comments (0)