
Remember me
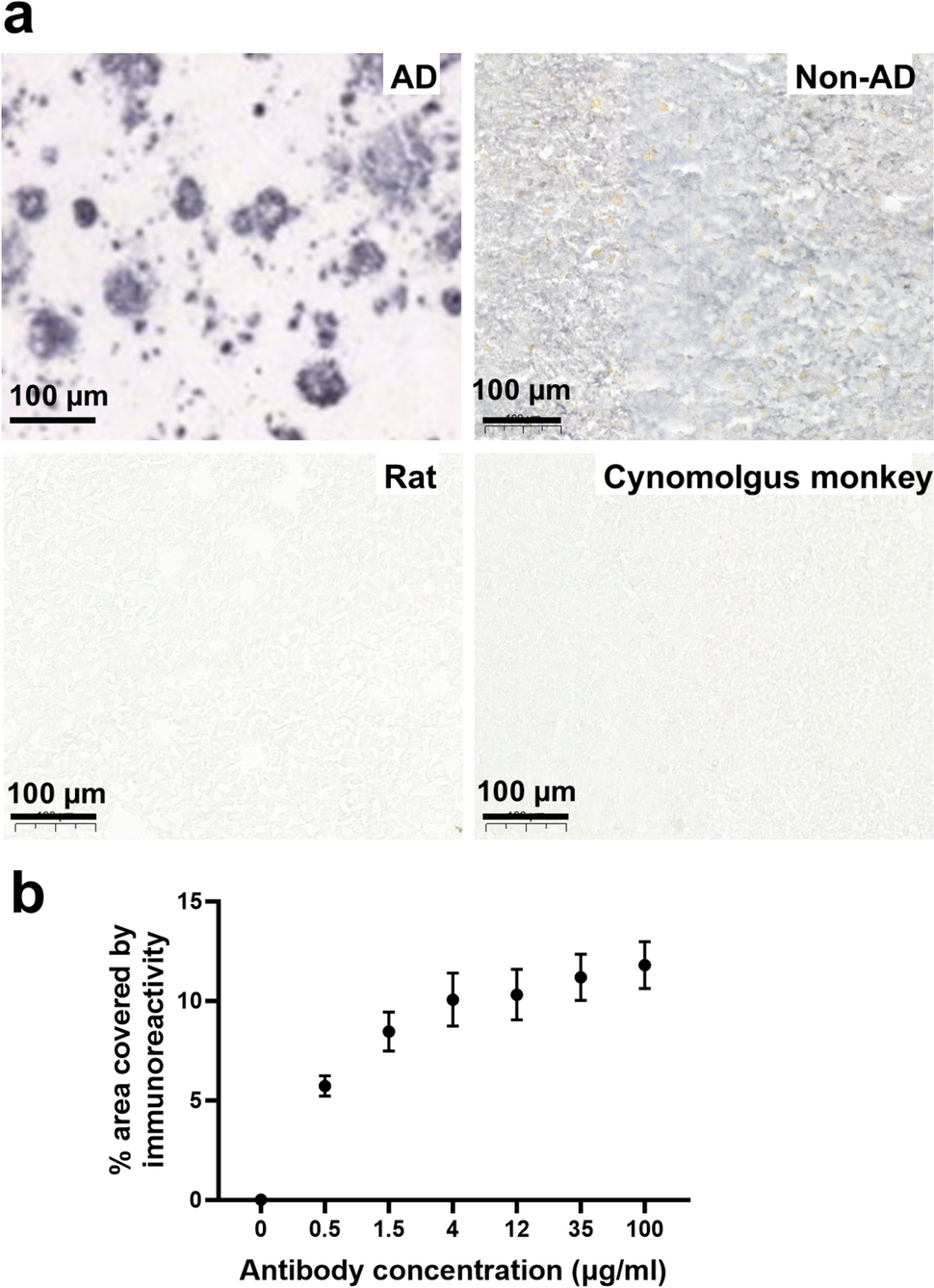

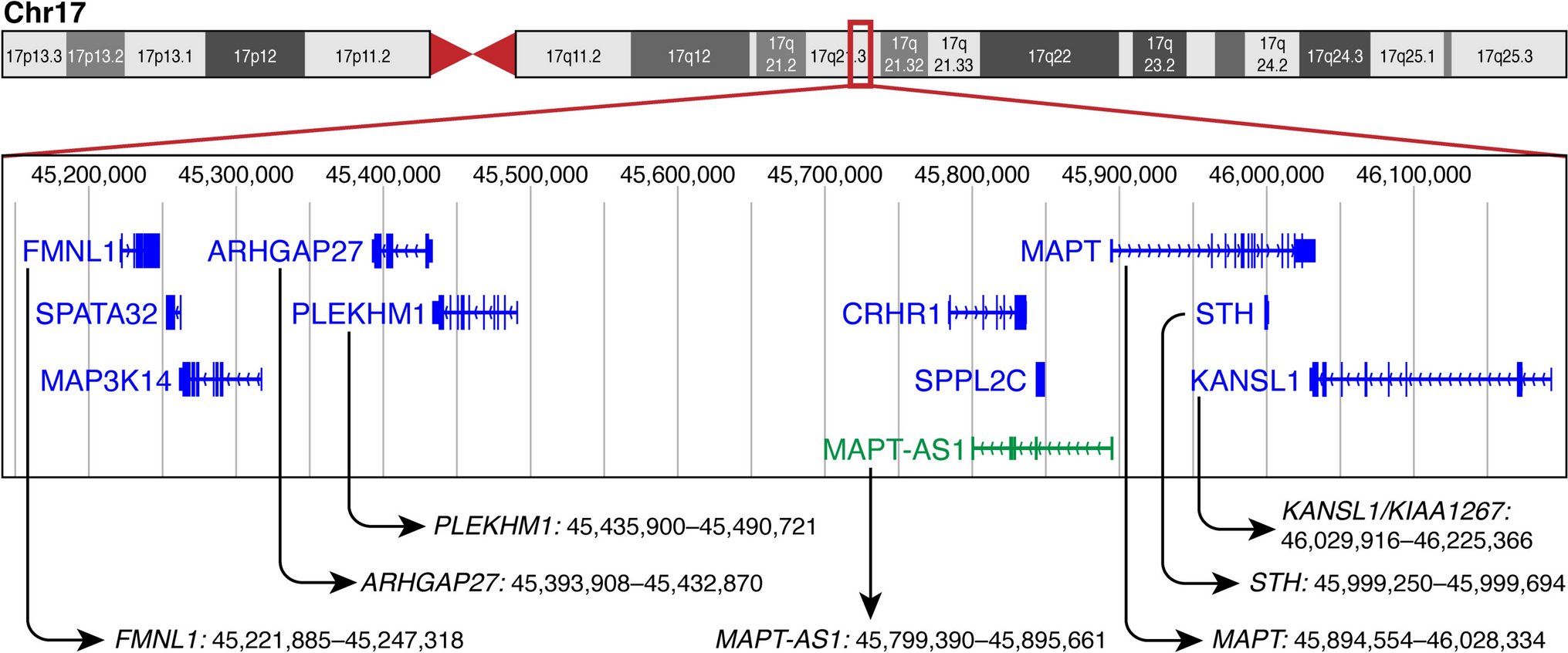
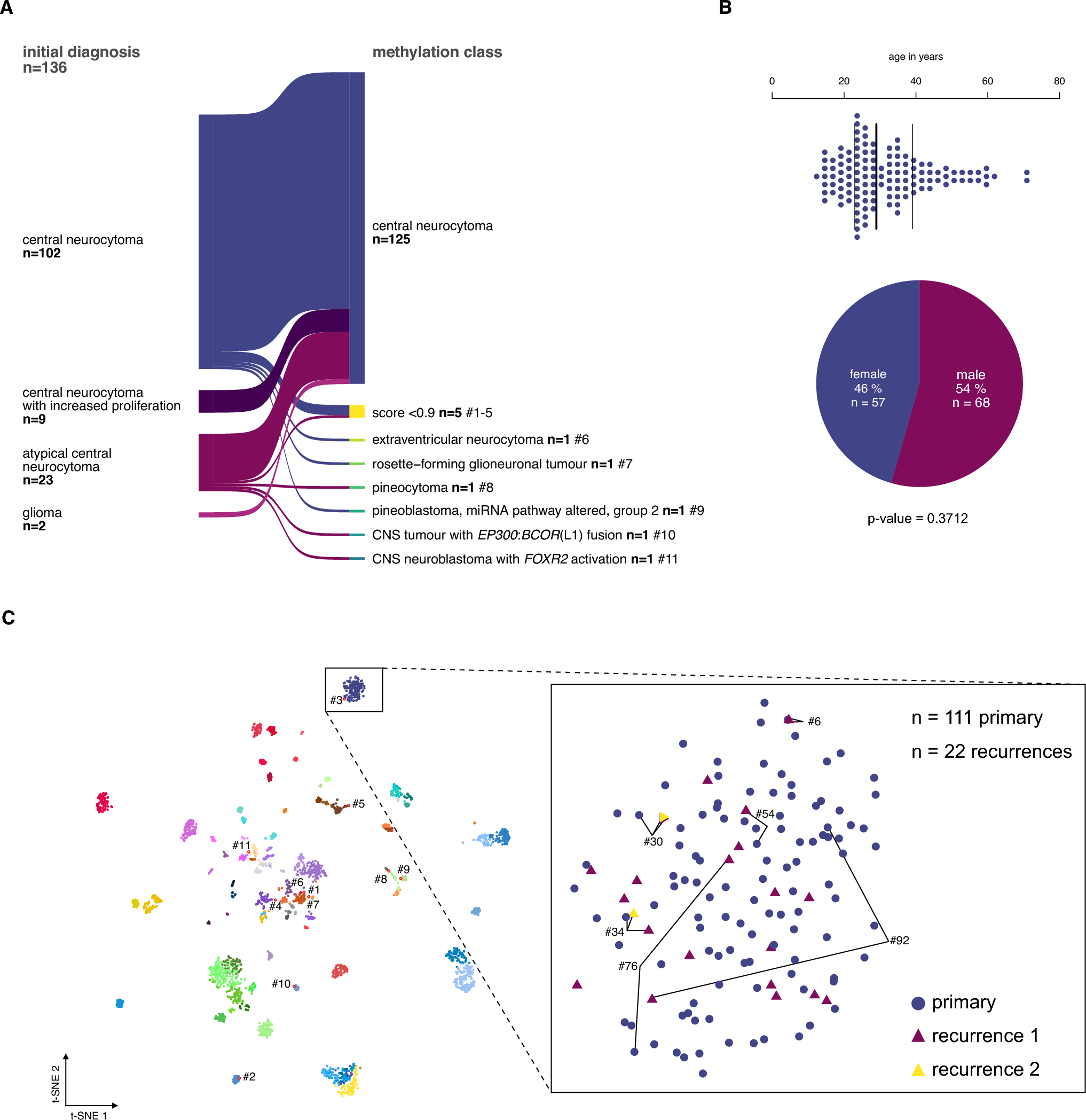



Sixty-five patients (40 males), including 48 with APT and 17 with PC, who were diagnosed and treated at specialized tertiary pituitary centers in 11 European countries (Belgium, Croatia, Denmark, Finland, Germany, Italy, Norway, Poland, Serbia, Sweden, and the UK) were enrolled in the study. A subset of 28 patients enrolled in this study was included in our previous study on ATRX gene expression in pituitary tumors [8]; no data on epigenetic alterations in any of the patients included in the present cohort have been published previously. The inclusion criteria were adult age (> 18 years) and strictly fulfilling the criteria for APT according to the ESE guidelines [33]: invasive tumor with unusually fast growth and/or clinically significant tumor progression despite surgery, radiotherapy, and standard medical therapy. In the PC group, seven patients presented with metastases within the central nervous system, seven had metastases to distant locations, and three had metastases in both the central nervous system and other sites. None of the patients had known metastases at the time of their first pituitary surgery. One patient was excluded as an outlier, as detailed below, resulting in a final cohort of 64 patients (39 males), comprising 48 with APT and 16 with PC.
The tumors were clinically classified based on the symptoms and results of the laboratory tests. Of the 64 APT/PC, 14 were clinically non-functioning. Among the 50 hormone-producing APT/PC, corticotroph and lactotroph tumors were the most common. Four patients presented with tumors with functional status that changed during the course of the disease; three silent corticotroph tumors developed into ACTH-secreting tumors, and one prolactin-secreting tumor changed to a growth hormone-secreting tumor. These patients were categorized into the Cushing’s and acromegaly groups, respectively. The specimens analyzed were obtained from surgeries performed before the phenotype changes.
A group of 12 patients (six males) who underwent surgery for a non-invasive PitNET (Knosp grades 0–2) at the Department of Neurosurgery, Uppsala University Hospital, was included for comparison. These patients had no signs of tumor regrowth or hormonal hypersecretion during ≥ 5 years of follow-up after surgery with or without standard pharmacological therapy. In the histological specimens, there was only slight cell atypia, < 2 mitoses per 10 high power fields (HPF; 1 HPF corresponding to 0.238 mm2), a Ki67 index < 3% and p53 expression in < 10 cells per 10 HPF.
The distribution of the histological and clinical tumor types in the cohort is presented in Table 1. Details regarding the origin of all histological specimens analyzed in the study, including both APT/PC and benign tumors, are outlined in Fig. 1.
Table 1 Overview over the entire cohortFig. 1
Chart showing the structure of the APT/PC cohort and the benign tumors
In the first surgery cohort (38 APT/PC and 12 patients with benign PitNETs), information on tumor invasion as assessed by magnetic resonance imaging (MRI) was available for 47 patients.
The Swedish Ethical Review Authority approved the study protocol, Dnr 2021-00073. PitNET tissue specimens from patients who underwent surgery at Uppsala University Hospital were partly obtained through the U-CAN project (www.u-can.uu.se) [13] and used in accordance with the ethical permission Dnr 2018–053.
Tissue specimensFormalin-fixed paraffin-embedded (FFPE) tumor tissue specimens were collected from the pathology departments at the participating centers. Hematoxylin–eosin-stained slides from all tissue specimens were reviewed by two pathologists (OC-B and JJ) to confirm the presence of representative tumor tissue and assess the fraction of viable tumor cells. In cases where a tissue sample contained both neoplastic and non-neoplastic pituitary tissue, the tumor tissue was macro-dissected for DNA extraction.
Tumor tissue specimens from multiple operations were available from 39 patients. When possible, specimens with the most representative and well-preserved tumor tissue from an early surgery, before radiotherapy or any pharmacological therapy, were selected for methylation profiling when possible. The first surgery specimens were excluded if they were too small or if the tissue was damaged (e.g., due to bleeding, ischemic damage, or damage from diathermia). In some cases, specimens could not be obtained. In five patients, for whom the intervals between pituitary surgeries were longer than 9 years or for whom material from the pituitary surgery as well as from the metastasis was available, two tumor tissue specimens were examined. This resulted in the analysis of a total of 69 APT/PC specimens (Fig. 1). Among the 16 patients with pituitary carcinoma, tissues from the first surgery were examined in seven cases, while tissues from recurrences were analyzed in seven cases (two of whom had paired specimens from pituitary surgery and metastasis). In two patients, tissues were only available from metastases. Seven of the nine patients with PC had received adjuvant treatment, including radiotherapy (n = 3), or radiotherapy and temozolomide (n = 4; one of these also received bevacizumab) before the specimens from recurrence/metastasis were obtained.
Histological classification was based on the immunohistochemical expression of anterior pituitary hormones (FSH, LH, TSH, ACTH, GH, and PRL) in tumor cells. Immunohistochemical (IHC) analyses were performed at the participating centers according to the routine diagnostic protocols. Additional IHC analyses of pituitary-specific transcription factors PIT1 (Novus Cat# NBP1-92,273, RRID: AB_11030310), TPIT (Atlas Antibodies Cat# AMAb91409, RRID: AB_2716678), and SF1 (Abcam Cat# ab217317, RRID: AB_2920891) were performed for cases of non-functioning (NF)-PitNETs that were immunohistochemically negative for all anterior pituitary hormones. For the first surgery specimens, IHC was also performed using monoclonal antibodies against Ki67 (clone MIB1, Agilent Cat# GA626, RRID: AB_2687921) and p53 (clone DO-7, Agilent Cat# GA616, RRID: AB_2889978) at the Department of Clinical Pathology, Uppsala University Hospital, according to the routine diagnostic protocols (p53 not performed in three cases, and Ki67 was not performed in one due to sparse tumor tissue in the FFPE specimens). Two pathologists (JJ and OC-B) independently quantified the Ki67 stainings and the p53 expression with a high concordance. Ki67 was quantified as the percentage of distinctly positive cells among 2,000 tumor cells (focusing on hotspots if present), while p53 was quantified as the total number of distinctly p53 positive cells in 10 HPFs.
Forty six of the fifty tumors from the first surgery cohort could be classified according to the French 5-tiered pituitary tumor classification [45].
DNA purificationDNA was purified from 10 μm-thick paraffin slides using the GeneRead DNA FFPE Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
Methylation profiling, tumor classification, and copy-number variation (CNV) analysisMethylation data were collected using the Illumina InfiniumMethylation EPIC (850 k) BeadChip Kit (Illumina, San Diego, CA, USA) as previously described [26, 32]. Bisulfite conversion was performed using the Zymo EZ DNA Methylation Kit (Zymo Research, Irvine, CA, U.S.A.), and bisulfite-converted DNA was restored using the Infinium HD FFPE Restore Kit from Illumina. The EPIC BeadChip array was scanned with the iScan from Illumina. IDAT files were uploaded to the MolecularNeuropathology.org server (https://www.molecularneuropathology.org/mnp) for in silico tumor classification and generation of copy-number variation (CNV) plots using workflows v11b4, v12.5, and v12.8, as previously described [6, 7].
CNV profiles calculated from methylation profiling data were obtained using the CNV script files embedded as part of the most recent brain tumor classifier workflow (v12.8). Overall CNV profiles for sample groups were generated by annotating individual CNVs in a cumulative manner, using at least 50% gain or loss for a given chromosomal arm as a cutoff for recording the number of chromosomal arm-level alterations for each sample. CNVs for each specimen were scored as gains, losses, or balances (indicating no events), and converted into a factorized matrix as input for hierarchical clustering of the associated CNV events based on the Jaccard similarity index distance. These distances were calculated using the ade4 R package [11] and visualized in a heatmap using the ComplexHeatmap R package [14, 15].
Fluorescence in situ hybridization (FISH)To validate the CNV data derived from the methylation analysis, we performed FISH analysis on 24 specimens, including 21 APT/PC and 3 benign PitNETs, using 2 µm sections from FFPE tissue blocks. Specimens were selected to represent the distinct EPIC-derived copy-number profiles among the APT/PC group. Probe sets XL BCR/ABL1 plus and XL DLEU/LAMP/12cen (MetaSystems Probes, Germany) were used to evaluate the ploidy states of chromosomes 9 and 22, as well as 12 and 13, respectively. Following a melting step at 85 °C for 10 min, probes were hybridized at 37 °C for 20 h using VYSIS HYBrite system. The sections were then diluted in stringency buffer, washed, and dehydrated using ethanol. Analysis was performed with VSViewer (MetaSystems, Germany). For each specimen and probe set, at least ten fields of interest were analyzed.
Data preprocessingWe analyzed the methylation array data in the R (v 4.3.2) environment using the ChAMP R package (v 2.32.0) [28, 44]. First, we imported the raw iDAT files and the sample sheet using the champ.load function. We then set the default quality control parameters and filtered out probes with overlapping SNP positions, sex chromosomes, and detection p values > 0.01. Next, we normalized the retained probes using the beta mixture quantile dilation method (BMIQ). For further analysis, we considered only the shared CpGs across the 81 specimens, leading to the selection of 384,629 CpG probes from an initial pool of 865,918 probes. Thereafter, we used the champ.SVD function to assess correlations between the covariates, including slide, array, sex, and age, and the principal components accounting for most of the variation in the data. This analysis revealed correlations between slide, array, and sex and the major principal components. To counteract these batch effects, we used the champ.runCombat function for batch correction. This procedure was also applied to analyze of the subset of 50 first surgery tumor specimens.
Unsupervised clustering and differential methylation analysisWe used the CpG probes with the top 5000 most variable β values (degree of methylation) as input for unsupervised hierarchical clustering. Clustering was based on the complete linkage method, and Pearson correlation was used as the distance method. The clustering and β values were visualized in a heatmap using the Heatmap function embedded in the ComplexHeatmap R package [14, 15]. Furthermore, we utilized the top 5,000 most variable β values as input for three-dimensional unsupervised clustering using principal component analysis (PCA) and t-distributed Stochastic Neighbor Embedding (tSNE), using the stats, rgl, and tSNE R-packages, respectively.
Differential methylation analysis was based on the β values and using the champ. The DMP function considered CpG probes with a nominal p value ≤ 1.3 × 10–7 (corresponding to genome-wide significance) and an absolute change in β value ≥ 0.2 as significantly methylated. Initial analysis of the shared CpGs from 83 specimens (including 65 APT/PC patients and 12 control patients) revealed two specimens as clear outliers. One was the only available specimen from a metastasis in a patient with a silent corticotroph tumor who also had two additional neoplasms: a rare retroperitoneal liposarcoma and colorectal adenocarcinoma, potentially indicating an underlying genetic tumor syndrome. The morphological appearance of this tumor was consistent with a neuroendocrine tumor, and there was distinct TPIT nuclear staining in the tumor cells. This patient was subsequently excluded from the cohort. The other outlier was a paired specimen from a tumor recurrence of a silent PIT1-positive APT, with no other known malignancy or syndrome. The morphological appearance, characterized by lobular architecture and moderate cell atypia, could suggest an immature PIT1-cell lineage tumor. However, the similarity between the first and recurrent specimens argues against dedifferentiation to an immature PIT1-cell lineage tumor over time. The recurrent specimen, which exhibited outlier behavior, was excluded, while the first surgery specimen was further analyzed as part of the first surgery cohort (Fig. 1). For information regarding the methylation class of the two outliers, see the Methylation profiling and tumor classification section in the Results section. The two outlier specimens were not part of the study cohort presented in Table 1.
We compared the degree of methylation between the APT/PC group and the benign tumor group, both in the entire dataset and in the subset of 50 tumor specimens obtained from the first surgery.
Gene set enrichment analysis of differentially methylated positions between aggressive and metastatic PitNETsGene set enrichment analysis (GSEA) was performed using the msigdbr and fgsea R packages. For each comparison of differential methylation, the CpGs were ranked by their β-values, from most positive to most negative. Next, CpGs located outside a gene loci were filtered out. For those CpGs located in multiple positions within the same gene, only the entry with the highest absolute value was kept for further analysis. The final pre-ranked individual genes were then subjected to GSEA. We used collections of human KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/) defined gene sets as input for the analysis. GSEA was conducted using 1000 permutations, with the eps set to zero. The minimum and maximum gene set sizes were set to 15 and 500, respectively. Gene sets with FDR ≤ 0.05 were considered as being statistically significantly enriched.
Comments (0)