
Remember me




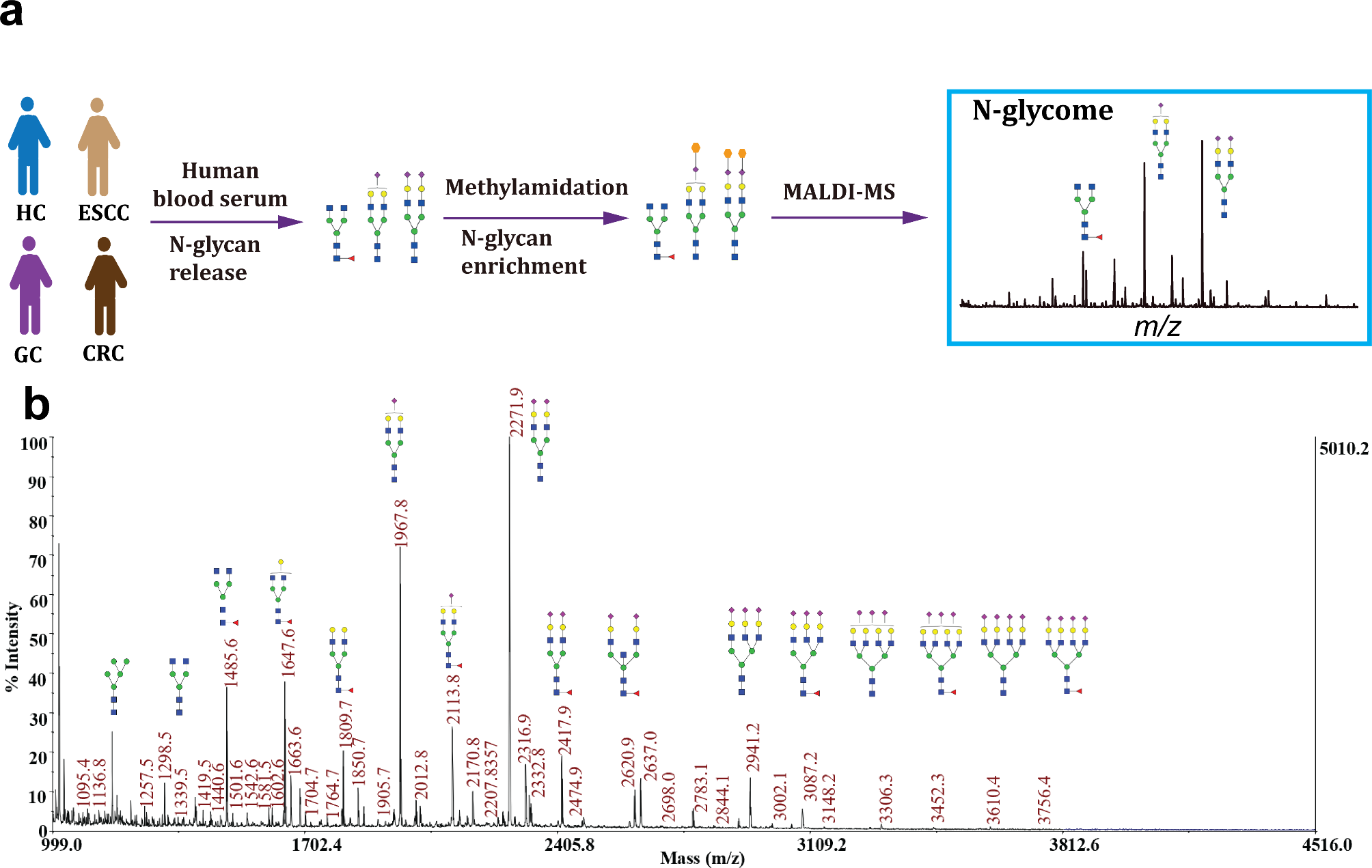
SPOT is designed to provide quantitative deep profiling of spatially-resolved proteomics by direct TMT labeling tissue proteins on slides (Fig. 1). Isobaric labeling with TMT is used as a proof-of-principle study, TMT is a well-established robust system [45] for multiplex, relative protein quantitation of up to 18 samples. TMT binds to primary amines (N-terminal and epsilon amino group of lysine residues) in proteins/peptides using NHS chemistry [46]. Particularly, in protein labeling, TMT can be conjugated to accessible lysine residues and the protein N-terminus. SPOT utilizes TMT for the controlled labeling of proteins spatially distributed on a 2D-tissue slide. Subsequently, the entire tissue section would be harvested and subject to standard proteomic analysis workflow.
Fig. 1
Overview of the SPOT workflow. Tissue slides are first annotated by cell types, histological patterns, or pathological states, followed by applying TMT tags directly onto regions of interest. After on-slide labeling and quenching of TMT, the tissue would be lysed, digested, and cleaned up for the downstream proteomic analysis using a mass spectrometer
TMT labeling at the protein-level on different tissue slides.The efficacy of direct TMT labeling at the protein-level was evaluated across various slide types, including frozen tissues without staining, FFPE without staining, deparaffinized FFPE, deparaffinized and decrosslinked FFPE, and tissues with H&E staining, H staining, E staining. A mixture of 18 TMT tags was directly applied to the tissue sections, a pipette was used in this study as the initial applicator for tissue labeling. Subsequently, the entire tissue slide was scraped off, lysed, digested, and cleaned up using SCX followed by C18 STAGE tips and MS analysis. Upon completion of MS data generation, the raw data of each slide was searched and evaluated based on the identifications at PSM, peptide, and protein levels, as detailed in Table S4.
In general, tissue proteins on various types of slides could be labeled with TMT, with varying degrees of labeled protein percentages. Across all types of tissue slides, only 10% or less of the total PSMs were identified to have TMT tags at the protein N-termini. Consistency could be observed for the identifications at PSM, peptide, and protein levels between the two repeats of the same tissue slide type (Table S4). Frozen sagittal mouse brain slides served as a reference for assessing labeling efficiency, given that minimal treatment was applied to frozen slides. In comparison to frozen slides, where TMT labels were found on over 92% of proteins, a visible decrease in the percentage of labeled proteins could be observed in untreated, deparaffinized, and deparaffinized/decrosslinked slides (Fig. 2A). Remarkably, the labeling of proteins (~ 64%) is minimally affected by the presence of paraffin compared to the deparaffinized ones (~ 70%), supposedly attributable to the permeability of acetonitrile through the paraffin. Paraffin-coated surface also has low surface energy (high contact angle over 100°) [47], which limits the lateral spreading of the TMT solution. An adept control of TMT dot size is critical in ensuring precise and reproducible labeling efficiency in more precise settings such as labeling on tissue microarray (TMA) slides (coring size ~ 0.6 mm). In addition, the decrosslinking step also improved the labeling efficiency by ~ 8%.
Fig. 2
Total identified proteins from each type of sagittal mouse brain slide. A Protein identifications of frozen, untreated FFPE, deparaffinized FFPE, and deparaffinized/decrosslinked slides (all were unstained). B Protein identifications of frozen, H&E stained, H stained, and E stained slides
Next, the effects of histology staining on direct tissue protein labeling were also evaluated (Fig. 2B). Notably, TMT tags were identified on over 92% of proteins from H-stained tissue slides. H&E stained (88%) and E-stained (84%) tissue slides showed a slightly lower percentage of labeled proteins compared to frozen and H-stained, however, the difference was less than 10%. During the H&E staining procedure, haemalum (oxidized hematoxylin solution) attaches to cell nuclei through covalent bonds between DNA phosphate oxygens and aluminum atoms, as well as between aluminum atoms and haemalum molecules. This covalent interaction between DNA and haemalum might release certain proteins bound to DNA, potentially elucidating the observed increase in protein identification with H-staining only. In contrast, eosin is attracted to tissue proteins by ionic forces (van der Waals forces) [48], and it could form salts with basic compounds like proteins. In turn, the presence of eosin could take up some of the binding capacity of SCX and C18 materials due to the hydrophobic interactions, providing a plausible explanation for the observed decrease in labeled protein percentage in H&E and E-stained slides.
On-site labeling of proteins from different brain regions on mouse brain slideFollowing the successful validation of direct labeling of tissue proteins on slides, we further evaluated SPOT's ability to detect proteomic patterns within a spatial context. A horizontal mouse brain slide with eight different regions was clearly outlined and each region was “stained” with a different TMT tag as illustrated in Fig. 3A. To enhance the visual recognition of the brain regions, the horizontal mouse brain slide was first stained with hematoxylin only, since hematoxylin did not interfere with the direct TMT labeling of tissue proteins (Fig. 2).
Fig. 3
A Mouse brain slide in horizontal view. Eight different regions are color-coded as shown and a scale bar to show the size of the brain slide. The scanning image was augmented using the filter “Hematoxylin” and brain regions were marked using QuPath52 [49]. B Hierarchical clustering illustrating the proteomic quantification results across 8 brain regions. Protein expressions could be clustered into 8 clusters, each revealing a distinctive spatial trend displayed on the left side of the heatmap
The mouse brain tissue was prepared similarly for downstream quantitative proteomics evaluation. Each region displayed a distinctive protein expression pattern and eight protein clusters were established using a soft clustering algorithm [50] (Fig. 3B, Figure S1, and Table S5). Among the eight clusters, cluster 4 (C4) and cluster 6 (C6) had obvious upward and downward protein expression trends starting from the neocortex to the cerebellum, respectively (Figs. 3B and S1). Excitatory amino acid transporter 1 (Eaa1), a glutamate transporter localized in the brain, was identified from C4 with the highest abundance in the cerebellum compared to the other regions, correlated well with a previous study showing that Eaa1 was highly enriched in the Purkinje cell layer in cerebellum [51]. On the other hand, elevated protein expression of V-type proton ATPase subunit a1 (Vpp1) in the neocortex was observed in C6. Vpp1 is reported to be predominantly expressed in neurons in the cortex and the dentate gyrus, part of the hippocampus. It can be found at low levels in astrocytes, oligodendrocytes, and microglia [52].
The results demonstrate that SPOT effectively detected proteomic patterns directly from tissue protein labeling indicating that SPOT is useful in studying spatial proteomics.
On-site labeling of different Gleason score regions on the frozen slide and TMA slideProstate cancer frozen slideTo further test the on-site labeling on frozen tissue slides in discerning smaller regions of interest, an experienced pathologist annotated 4 regions of 0.6 mm in diameter within normal sections, Gleason score 3 sections, Gleason score 4 sections, and Gleason score 5 sections, respectively (Figs. 4A). Based on the pathological annotations on the adjacent H&E slides, direct TMT labeling was carried out on the frozen slides.
Fig. 4
On-site TMT labeled frozen prostate cancer tissue slide. A Bright-field scanning of the adjacent prostate cancer H&E slide annotated with normal (yellow), Gleason 3 (cyan), Gleason 4 (blue), and Gleason 5 (purple) regions. B Principal component analysis of Gleason score regions based on the protein expression profiles. C Hierarchical clustering based on the expression profiles of 289 proteins across different Gleason score regions. D Significantly changed proteins (absolute log2 fold change > 1, p-value < 0.05) from pairwise comparison of two different Gleason score regions
In total, 11,214 peptides were identified, corresponding to a set of 1,854 unique proteins. Within this dataset, 1,365 peptides were successfully labeled with TMT tags, corresponding to 289 distinct proteins. Following this identification, hierarchical clustering and principal component analysis (PCA) and hierarchical clustering were conducted to examine the association among different Gleason score regions based on their protein expression profiles (Fig. 4A, B). Notably, normal regions and Gleason4 regions could be separated completely, whereas Gleason 3 and Gleason 5 regions had a considerable overlap. Regions characterized by normal or the same Gleason score (ranging from Gleason 3 to 5) displayed notably high correlations across diverse tissue sections (Figure S2A). Conversely, regions associated with different Gleason scores exhibited relatively lower degrees of correlation.
Furthermore, differential analysis (Fig. 4D) was able to return two proteins specifically enriched in the prostate tissue (Human Proteome Atlas [2, 43]), two proteins that were found to relate to poor prognosis of prostate cancer (Human Proteome Atlas [2, 43]), and three proteins related to cell markers in the prostate (Cell Marker 2.0) [44]. Previous studies have indicated notable clinical relevance associated with microseminoprotein-beta (MSMB) [53] and epithelial cell adhesion molecule (EPCAM) [54, 55] for prostate cancer. In this study, MSMB was found to be overexpressed in Gleason 3 regions relative to normal regions, while EPCAM was found to be elevated in both Gleason 3 and Gleason 5 regions, but higher fold change was observed in Gleason 3 compared to normal (log2 fold change = 1.75) than Gleason 5 compared to Gleason 4 (log2 fold change = 1.08) (Fig. 4D). MSMB and EPCAM could be prostate cancer-relevant indicators or contributors in various medical and pathological conditions, underscoring the importance of further exploration and validation.
In summary, these results indicate that SPOT could capture potential correlations and variations in molecular profiles across different Gleason scores from frozen tissue slides even with direct TMT labeling of proteins in the 0.6 mm region.
Prostate cancer TMA slideFollowing the application of TMT direct labeling onto frozen tissue slides derived from prostate cancer specimens, there arises a distinct interest in evaluating the translatability and consistency of this labeling methodology when extended to TMA slides. TMA cores were meticulously assessed by an experienced pathologist who assigned distinct scores to each core, based on the H&E stained adjacent slide (Fig. 5A). Eighteen cores of the size 0.6 mm were selected for TMT direct labeling (three for normal, five for Gleason score 3, five for Gleason score 4, and five for Gleason score 5).
Fig. 5
On-site TMT labeled prostate cancer TMA slide with paraffin. A Bright-field scanning image of selected cores from the adjacent H&E TMA of prostate cancer. Three normal cores, five Gleason score 3 cores, five Gleason score 4 cores and five Gleason score 5 cores were represented. B PCA analysis based on the protein expression profiles in different Gleason score regions. C Hierarchical clustering using the expression profiles of 265 proteins across different Gleason score regions. D Significantly changed proteins (absolute log2 fold change > 1, p-value < 0.05) from pairwise comparison of two different Gleason score cores
The TMA format involves the systematic arrangement of discrete tissue cores, evenly spaced across the slide, providing a representative sampling of various specimens. Importantly, the deliberate spacing of these cores minimizes the risk of label mixing between different regions, ensuring a more accurate and region-specific evaluation of the TMT labeling method within the TMA framework. This comparative analysis aims to contribute valuable insights into the method's adaptability and reliability across different tissue slide formats, advancing our understanding of its applicability in broader histological contexts.
In total, 1,873 peptides (corresponding to 560 unique proteins) were identified, out of which 790 were TMT-labeled peptide sequences. These labeled peptides corresponded to 265 distinct proteins. Principal component analysis revealed minimal to no overlaps between each group (Fig. 5B), indicating distinct clustering patterns. The subsequent hierarchical clustering analysis (Fig. 5C) provided additional insight, revealing varying degrees of mixing between each group. This observation implies nuanced relationships and molecular heterogeneity within the regions characterized by different Gleason patterns.
Furthermore, the TMA dataset identified four proteins (PTMA, PPAP, POSTN, AGR2) that exhibited potential in distinguishing different Gleason score regions (Gleason score 3, 4, and 5). Additionally, one cell marker protein (MYH11) demonstrated the capability to differentiate normal regions from different Gleason score regions (Fig. 5C), a finding consistent with observations in the frozen dataset. These proteins identified in the TMA dataset, particularly the four proteins demonstrating variations among different Gleason score regions, suggest their potential for clinical applications in prostate cancer detection. Their implications in prostate cancer, as reported in previous studies [56,57,58,59,60,61,62,63,64], further underscore the significance of these proteins in the context of prostate cancer pathology. Understanding the molecular basis of Gleason patterns through these proteins could enhance the precision of prostate cancer grading. In addition, the cell marker protein capable of distinguishing normal from cancerous regions holds diagnostic potential and may serve as a valuable tool for clinicians in accurately identifying cancerous areas within the prostate.
Based on the correlation analysis result (Figure S2B), a pronounced association could be observed within normal cores as well as within Gleason score 3 cores. This strong correlation suggests a potential consistency in molecular characteristics within normal tissues and within low-grade prostate cancer. In contrast, the correlations observed among Gleason score 4 and Gleason score 5 cores exhibit greater variability, suggesting potential differences in tumor heterogeneity. The varied correlations observed in the higher-grade cores suggest the intricate nature of prostate tumor heterogeneity [56, 65]. This complexity arises from differences in the types and arrangements of cells, which can influence unique molecular characteristics within the tumor.
Discussion and future directionsThis study demonstrates a new approach, SPOT, for studying spatial proteomics quantitatively via direct labeling on tissue slides coupled with bottom-up MS. We were able to characterize different mouse brain regions as well as regions of different pathological states of prostate cancer directly from tissue slides of various forms by using SPOT. While these results are promising, further validation and optimization are necessary to fully exploit the potential of TMT labels for tissue slide analysis.
Applying labels directly onto tissue slides offers several advantages, including the ability to multiplex and analyze multiple samples in depth simultaneously, providing a comprehensive understanding of the tissue’s molecular landscape. Applying TMT directly onto proteins can present certain challenges. For instance, TMT can obstruct trypsin cleavage at lysine residues, resulting in longer peptides. To mitigate the effects from TMT protein-level labeling, utilization of alternative proteases may improve identification rate and coverage. Labeling at protein-level also grants us limited access to labeling sites, unlike peptide-level labeling, where TMT can easily target lysine residues and N-terminal sites. A peptide-level SPOT approach could overcome this limitation by applying a protease before TMT labeling. Additionally, SPOT was only applied to regions of interest, leaving most of the tissue slice unlabeled. We could improve the labeling efficiency by labeling the areas of interest with specific TMT channels in situ, then collecting the tissue and further labeling the samples with a different TMT tag in solution. Nonetheless, SPOT enables labeling the protein in its original context, enhancing the accuracy and relevance of the analysis.
As demonstrated using mouse brain and prostate cancer tissue slides, quantitatively profiling spatially distributed proteomes was achieved using SPOT. In the case of prostate cancer, the inherent heterogeneity of prostate tumors, characterized by diverse cellular populations and architectural patterns, poses a challenge for conventional Gleason scoring. While Gleason scoring remains a cornerstone in prostate cancer pathology, its limitations in fully capturing the intricacies of heterogeneous tumors are acknowledged. Ongoing research endeavors are dedicated to refining grading systems [66], particularly for tumors displaying mixed patterns, or using artificial intelligence-aided diagnosis [67, 68]. Our technology SPOT offers a viable solution for multiplexed spatial profiling using bottom-up proteomics and has the potential to unveil the complexities of prostate cancer heterogeneity. However, the current resolution of SPOT is insufficient to capture localized heterogeneity at the single-cell level. To address this limitation, exploring robust tissue labeling methods that offer enhanced precision and reproducibility is undoubtedly the next step. Techniques such as machine learning-assisted imaging processing and automated robotic arms equipped with high-precision imaging systems that can perform precise tissue manipulation and labeling may be considered. Future studies should also involve a larger sample size and include a broader range of pathological states to validate the observed differences and confirm the specificity and clinical utilities of the identified proteins.
Besides quantitatively profiling spatially distributed proteomes, we envision utilizing SPOT for the identification of protein–protein interactions and interactions with other binding partners (such as DNA, RNA, and metabolite) within specific subcellular compartments. Perturbations in the cellular microenvironment could induce alterations in the natural patterns of these interactions [69]. Such changes may arise as a consequence of environmental stressors or disease conditions, influencing the intricate network of interactions that govern cellular responses and functions. Identification of protein-binding partners at a spatial resolution is instrumental in deciphering not only the functionality of individual proteins but also the intricate protein pathways involved in biological and pathological processes.
In addition, the integration of spatial proteomics data with other omics data, such as genomics, transcriptomics, and metabolomics, can provide a comprehensive understanding of cellular processes and disease mechanisms. For example, the integration of spatial proteomics data with transcriptomics data can provide insight into the regulation of protein localization and expression, while the integration of spatial proteomics data with metabolomics data can provide insight into the functional consequences of alterations in protein localization or expression. As spatial transcriptomics advances towards unraveling the spatiotemporal intricacies of gene regulation [70], a similar perspective could be applied to spatial proteomics. In contrast to spatiotemporal transcriptomics, spatiotemporal proteomics has the potential to directly reveal the consequences of gene expression alterations across both spatial and temporal dimensions.
Comments (0)