
Remember me

The App single knock-in mouse model (AppNL-G-F; Takaomi Saido)47 does not overexpress APP as in classical APP mouse models, but contains the humanized Aβ sequence, as well as Swedish (NL), Arctic (G) and Iberian (F) mutations. AppNL-G-F mice accumulate Aβ plaques and suffer from learning, memory and attention impairments from 6 months onwards47,48. The humanized Apphu/hu mice (named AppWT in the main text) were recently generated in our laboratory to serve as controls27. Both strains were crossed with homozygous Rag2tm1.1FlvCsf1tm1(CSF1)FlvIl2rgtm1.1FlvApptm3.1Tcs mice (Jackson Laboratory, strain 017708) to generate the Rag2−/−Il2rγ−/−hCSF1KIAppNL-G-F and the Rag2−/−Il2rγ−/−hCSF1KIApphu/hu used in this study. In total, we transplanted >150,000 cells across 11 different cell lines, and three mouse background genotypes. For all experiments, we used Total-Seq A hashing antibodies (BioLegend) so as to be able to demultiplex individual mouse replicates (Supplementary Fig. 2). Mice had access to food and water ad libitum and were housed with a 14/10-h light/dark cycle at 21 °C and 32% humidity, in groups of 2–5 animals. All experiments were conducted according to protocols approved by the local Ethical Committee of Laboratory Animals of KU Leuven (Ethische Commissie Dierproeven project no. P177/2017) following country and European Union guidelines.
Generation of Rag2 −/−Il2rγ −/−hCSF1 KIApp NL-G-FApoE −/− miceApoE knockout mice were generated in zygotes from homozygous Rag2tm1.1FlvCsf1tm1(CSF1)FlvIl2rgtm1.1FlvApptm3.1Tcs mice using CRISPR–Cas9 technology by targeting exon 4 of the mouse ApoE gene. The RNA guide 5′-CCTCGTTGCGGTACTGCCCGAGT-3′ was selected using the CRISPOR web tool. Ribonucleoproteins containing 0.3 μM purified Cas9HiFi protein (Integrated DNA Technologies), 0.3 μM CRISPR RNA (crRNA) and 0.3 μM trans-activating crRNA (Integrated DNA Technologies) were injected into the pronucleus of 72 embryos by microinjection in the Mouse Expertise Unit of KU Leuven. Two candidate pups were identified by PCR analysis with several primer combinations. One founder was selected for breeding and an allele with a chromosomal deletion of 335 base pairs (bp) (corresponding to 148 bp of intronic sequence and the first 187 bp of exon 4 sequence) (Extended Data Fig. 4) was selected to establish the colony. The founder mouse was backcrossed over two generations before a homozygous colony was established, which was designated AppNL-G-FApoE−/−. The strain was maintained on the original C57Bl6J:BalbC background. Standard genotyping for the ApoE allele was performed by PCR with primers 5′-GCTCCCAAGTCACACAAGAA-3′ and 5′-CTCACGGATGGCACTCAC-3′, resulting in a 755-bp amplicon for the WT allele and a 420-bp amplicon for the ApoE knockout allele.
Differentiation of microglial progenitorsUKBIO11-A, BIONi010-C and H9-WA09 and their isogenic modifications (Table 1) were differentiated into microglial precursors and transplanted following our recently published protocol, MIGRATE26. In brief, stem cells were plated and maintained in human Matrigel-coated six-well plates and in E8 flex media until reaching ~70–80% confluence. Once confluent, stem cell colonies were dissociated into single cells and plated into U-bottom 96-well plates at a density of ~10,000 per well in mTeSR1 medium with BMP4 (50 ng ml−1), VEGF (50 ng ml−1) and SCF (20 ng ml−1) for 4 d. On day 4, embryoid bodies were transferred into six-well plates (~20 embryoid bodies per well) in X-VIVO (+supplements) medium supplemented with SCF (50 ng ml−1), M-CSF (50 ng ml−1), IL-3 (50 ng ml−1), FLT3 (50 ng ml−1) and TPO (5 ng ml−1) for 7 d with a full change of medium on day 8. On day 11, differentiation medium was replaced with X-VIVO (+supplements) with FLT3 (50 ng ml−1), M-CSF (50 ng ml−1) and GM-CSF (25 ng ml−1). On day 18, human microglial precursors were collected and engrafted into P4 mouse brains (0.5 million cells per pup) as previously described. Before transplantation, mouse microglia were depleted by inhibiting CSF1 receptor (CSF1R) with BLZ945 (dose of 200 mg kg−1) at P2 and P3 as previously described14,26. To confirm the biological activity of known essential mouse cytokines on human microglia, we cultured microglial progenitors in TIC medium49 for 9 d, supplemented with either hCSF1, hIL-34, hTGFb and hCX3XR1, or hCFS1, mIL-34, mTGFb and mCX3CR1, and measured the expression of several microglial makers by quantitative PCR (qPCR). We did not find any differences in the levels of P2RY12, CX3CR1, C1Q, HEXB, TGFbR1 and TREM2 (Supplementary Fig. 1a).
Genetic modification of stem cell linesGeneration of TREM2−/− and TREM2+/R47H from H9-WT (WA09) human embryonic stem cells was done as described by Claes et al.46. Briefly, the TREM2+/R47H CRISPR–Cas9 nickases and the two guide RNAs (gRNAs) (gRNA A and B) that target exon 2 of TREM2 nearby the location of R47H (G>A) and a genomic TTAA were purchased from Addgene. A donor plasmid was made, comprising homology arm 1 (HA1) of TREM2 (with the R47H mutation), a selection cassette (CAGG promoter, HYG/TK, green fluorescent protein) and HA2 of TREM2 exon 2. To create TREM2−/− human pluripotent stem cells (hPSCs), a CRISPR–Cas9, gRNA B and the same donor plasmid were used. To create TREM2+/R47H hPSCs, 2 × 106 single cells of the heterozygously targeted clone were nucleofected with 4 μg of piggyBac transposase plasmid and negative selection with fialuridine, also known as 1-(2-deoxy-2-fluoro-1-d-arabinofuranosyl)-5-iodouracil (FIAU) (1:8,000–1:2,500; 0.5 mM in water), was applied to select for cells wherein the selection cassette was removed. Of note, the H9-WT line from which the TREM2+/R47H and TREM2−/− lines were created carries an APOE ε3/ε4 genotype.
To get the gRNA into the cells, nucleofection was performed. Briefly, 2 × 106 single-cell H9s were preincubated with Revitacell (Life Technologies) and nucleofected using the Amaxa Nucleofector II on setting F16 with 2.5 μg of CRISPR–Cas9, 2.5 μg of gRNA (A and) B and 5 μg of donor template to create TREM2−/− hPSCs. Selection was initiated after 2–3 d with 25–150 μg ml−1 Hygromycin B (Sigma-Aldrich) and maintained for 10–15 d. Recombinant colonies were manually picked and expanded for further characterization.
gRNA A
F: CACCGACCCAGGGTATCGTCTGTGA
R: AAACTCACAGACGATACCCTGGGTC
gRNA B
F: CACCGCACTCTCACCATTACGCTG
R: AAACCAGCGTAATGGTGAGAGTGC
The H9 (WA09) embryonic stem cell line was also modified to express the CAS9 in a doxycycline-dependent manner (iCas9), using TALENS at the AAVS1 locus to facilitate the generation of targeted gene deletion. H9-iCas9 was used only in libraries 14 and 16 and doxycycline was never administered.
APOE expression using qPCRThe levels of APOE expression in the grafted stem cell lines were checked by collecting cell pellets. Using the RNeasy Micro Kit, RNA was extracted according to manufacturer’s instructions and the RNA was reverse transcribed with the High-Capacity cDNA Reverse Transcription Kit. A qPCR was performed with SensiFast SYBR reagent and custom-made primers for GAPDH (FW: tcaagaaggtggtgaagcagg; RV: accaggaaatgagcttgacaaa) and APOE (we average the level of expression of multiple primers spanning the whole gene, see table below).
hAPOE_1_F
F: TAGAAAGAGCTGGGACCCT
R: CACAGAACCTTCATCTTCCT
hAPOE_2_F
F: GTTGCTGGTCACATTCCTG
R: GCAGGTAATCCCAAAAGCGA
hAPOE_3_F
F: CTGGGTCGCTTTTGGGATTA
R: GTCAGTTGTTCCTCCAGTTC
hAPOE_4_F
F: AATCACTGAACGCCGAAG
R: TTATTAAACTAGGGTCCACC
hAPOE_qPCR_F
F: GTTGCTGGTCACATTCCTGG
R: GCAGGTAATCCCAAAAGCGAC
Soluble Aβ preparation and intracerebroventricular injectionsSoluble Aβ aggregates (10 μM) or scrambled peptides (10 μM) were prepared as previously14,50. Briefly, recombinant Aβ (1–42) or scrambled peptides were thawed during 30 min at room temperature and dissolved in hexafluoroisopropanol at 2 mg ml−1. Hexafluoroisopropanol was fully evaporated with a gentle stream of N2 gas and resulting peptides were dissolved in dimethylsulfoxide at 2 mg ml−1. Dimethylsulfoxide medium was removed using HiTrap Desalting column 5kD and peptides were eluted in Tris-EDTA buffer. Of note, Tris-EDTA buffer was composed of 50 mM Tris buffer and 1 mM EDTA at pH 7.5. Tris-EDTA-eluted Aβ or scrambled peptides were quantified using Bradford assay before aggregation. Peptides were left to aggregate for 2 h at room temperature. After 2 h, Aβ (1–42) or scrambled aggregates were diluted to a final concentration of 10 μM in Tris-EDTA buffer, snap frozen and stored at −80 °C. Following a similar approach as previously described14, at 12 weeks of age, AppWT mice engrafted with the full isogenic series of UKBIO11-A or BIONi010-C were anesthetized with isoflurane and injected intracerebroventricularly with 5 μl of soluble aggregates of Aβ (10 μM) or scrambled peptides (10 μM). Stereotactic coordinates from Bregma: anteroposterior: −0.22 mm; mediolateral: −1 mm; dorsoventral: −2.74 mm. After surgery, mice were placed on a thermal pad until recovery. At 6 h after injection, AppWT mice were euthanized, and human microglia were isolated using FACS for transcriptomics analysis.
Human microglia isolation from mouse brain for single-cell transcriptomicsAt 6–7 months of age AppNL-G-F, AppNL-G-FApoE−/− and AppWT mice xenotransplanted with H9, UKBIO11-A, BIONi010-C (Table 1) and their isogenic modifications were killed with an overdose of sodium pentobarbital and immediately perfused with ice-cold 1 × DPBS (Gibco, Cat. no. 14190-144) supplemented with 5 U of heparin (LEO). After perfusion, one hemisphere of each mouse brain without cerebellum and olfactory bulbs was placed in FACS buffer (1 × DPBS, 2% FCS and 2 mM EDTA) + 5 μM actinomycin D (ActD; Sigma, Cat. no. A1410-5MG) for transcriptomics. Brains were mechanically and enzymatically dissociated using Miltenyi Neural Tissue Dissociation Kit P (Miltenyi, Cat. no. 130-092-628) supplemented with 5 μM ActD. Next, samples were passed through a 70-μm strainer (BD2 Falcon), washed in 10 ml of ice-cold FACS buffer + 5 μM ActD and spun at 300g for 15 min at 4 °C. Note that 5 μM ActD was kept during collection and enzymatic dissociation of the tissue to prevent artificial activation of human microglia during the procedure as previously reported12. ActD was removed from the myelin removal step to prevent toxicity derived from long-term exposure. Following dissociation, myelin was removed by resuspending pelleted cells in 30% isotonic Percoll (GE Healthcare, Cat. no. 17-5445-02) and centrifuging at 300g for 15 min at 4 °C. Accumulating layers of myelin and cellular debris were discarded and Fc receptors were blocked in FcR blocking solution (1:10, Miltenyi, Cat. no. 130-092-575) in cold FACS buffer for 10 min at 4 °C. Next, cells were washed in 5 ml of FACS buffer and pelleted cells were incubated with the following antibodies: PE-Pan-CD11b (1:50, Miltenyi, Cat. no. 130-113-806), BV421-mCD45 (1:500, BD Biosciences, Cat. no. 563890), APC-hCD45 (1:50, BD Biosciences, Cat. no. 555485), Total-Seq A cell hashing antibodies (1:500, BioLegend) and viability dye (1:2,000, eFluor 780, Thermo Fisher Scientific, Cat. no. 65-0865-14), in cold FACS buffer during 30 min at 4 °C. After incubation, cells were washed, and the pellet was resuspended in 500 μl of FACS buffer and passed through a 35-μm strainer before sorting. For sorting, the cell suspension was loaded into the input chamber of a MACSQuant Tyto Cartridge and human cells were sorted based on CD11b and hCD45 expression at 4 °C (Supplementary Fig. 1). FACS data were analyzed using FCS Express 7 software.
HistologyWhen killing and collecting brains of mice for single-cell sequencing, one hemisphere of AppNL-G-F, AppNL-G-FApoE−/−and AppWT mice xenotransplanted with H9, UKBIO11-A and BIONi010-C was also preserved and postfixed in 4% PFA overnight at 4 °C. After 24 h, PFA was removed and they were washed and kept in 1 × DPBS at 4 °C until further processing. For sectioning, olfactory bulbs and cerebellum were discarded and brains were cut coronally (40-µM thickness) with a vibrating microtome (Leica). Each sample was collected under free-floating conditions in a series of six sections and stored in cryoprotectant solution (40% PBS, 30% ethylene glycol, 30% glycerol) at −20 °C. For staining, sections are washed in 1 × DPBS and permeabilized for 15 min at room temperature in PBS with 0.2% Triton. After permeabilization, sections were stained with X-34 staining solution (10 µM X-34 (Sigma-Aldrich), 20 mM NaOH (Sigma-Aldrich) and 40% ethanol) for 20 min at room temperature. Sections were washed several times with 40% ethanol for 2 min and with PBS + 0.2% Triton for 5 min. For the staining of microglia with anti-hP2RY12 (HPA014518, Sigma-Aldrich, 1:2,000), CD9 (312102, BioLegend, 1:100), FTH1 (PA5-1905, Invitrogen, 1:500) and HLA antibodies (ab7856, Abcam, 1:200), sections were blocked with 5% normal donkey serum in PBS + 0.2% Triton for 1 h at room temperature. For the costaining of CD9/FTH1 with P2RY12, primary antibody incubation was done overnight at 4 °C. For the HLA staining, signal was enhanced using a Tyramide SuperBoost kit (B40915, Thermo fisher) according to the manufacturer’s instructions. Briefly, after overnight incubation with HLA primary antibody, sections were incubated with a poly-HRP-conjugated secondary antibody. Tyramid solution was then added to the slices for 5 min and they were washed in PBS + 0.2% Triton after the reaction was stopped. The HLA sections were later costained with P2RY12 as previously described. Secondary antibodies were incubated for 1 h at room temperature. Finally, sections were mounted with Mowiol mounting medium (Sigma-Aldrich) or DAKO mounting medium (Agilent). Images at ×4 and ×20 magnification were taken on a Nikon A1R Eclipse confocal microscope. To measure the shift in microglial cell states at the site of Aβ plaques, we used a modified Sholl analysis where the fluorescent intensity of microglial markers P2RY12, CD9 and HLA was measured through concentric rings (annuli) of increasing diameter surrounding the X-34 plaque center. The analysis was performed using ImageJ software after determining a threshold for background correction. Intensities of each channel were scaled for comparison using z-score normalization. Intensity over distance (µm) was plotted using Loess nonparametric regression in R with estimated standard error for each predicted value. For comparison of intensities near and distant from the plaque center, the means of the inner and outer three annuli were independently calculated. Bar plots were generated in Prism v.10.
Single-cell library preparation and sequencingFor single-cell RNA sequencing, 15,000–20,000 human microglia (CD11b+, hCD45+) from each mouse were sorted on the MACSQuant Tyto (Supplementary Fig. 1) and diluted to a final concentration of 1,000 cells per µl. Since all the samples were individually hashed using Total-Seq A cell hashing antibodies, 2,000 human microglia per animal were pooled and loaded onto the Chromium Next GEM Chip G (PN no. 2000177). The DNA library preparations were generated following the manufacturer’s instructions (CG000204 Chromium Next GEM Single Cell 3′ Reagent Kits v3.1). In parallel, the hashtag oligo libraries were prepared according to the manufacturer’s instructions (BioLegend, Total-Seq A Antibodies and Cell Hashing with 10x Single Cell 3′ Reagent Kit v3 3.1 Protocol) using 16 cycles for the index PCR. A total of 20 libraries containing 95 biological replicates were sequenced, targeting a 90% messenger RNA and 10% hashtag oligo library (50,000 reads per cell), on a HiSeq4000 or NovaSeq6000 (Illumina) platform with the recommended read lengths by the 10X Genomics workflow.
Statistics and reproducibilityStatistical analysis of the distribution of different experimental groups across clusters was performed using each mouse as a single replicate. Normality and equal variance were tested, and the data were normalized if needed. We used both t-test and one-way analysis of variance (ANOVA) when comparing two or more than two groups, respectively. Statistical significance was set at P < 0.05, and multiple comparisons Bonferroni was applied when necessary. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications3,14. All the mice were randomized across the different experimental groups. Data collection and analysis were performed by blind researchers. Immunohistochemical data were repeated at least five times with similar results.
Analysis of single-cell RNA sequencing datasetsAlignment and softwareThe raw BCL files were demultiplexed and aligned by Cellranger (v.3.1.0) against the human genome database (hg19, Ensembl 87). Raw count matrices were imported in R (v.4.1.3) for data analysis. Datasets were analyzed using the Seurat R package pipeline (v.4.0.1). For specific statistical tests and visualizations, we also used GraphPad Prism v.9.0, Python, R and Bioconductor.
Quality control of cells and samplesFor each library included in this study, we excluded low-quality cells (poorly sequenced, damaged or dead cells) by filtering out cells with <1,000 reads or <100 genes detected. We also excluded cells with >15% of reads aligning to mitochondrial genes. Doublets were first excluded by removing cells with a number of reads or genes more than 3 s.d. from the library mean (Supplementary Fig. 2a). Doublets removal was further refined with cell hashing information by using Seurat’s function MULTIseqDemux() to assign cells to singlets, doublets or negatives (Supplementary Fig. 2b). Only singlets were retained, as negative cells cannot be demultiplexed and assigned with certainty to the sample of origin. For one library out of 20 sequenced for this study (library 9), the counts related to one hash (sample MG452) were high across all samples in the library and MULTIseqDemux() failed to demultiplex many cells. We used the function HTODemux() instead to demultiplex library 9 which performed better. Sample MG452 was entirely removed in further quality control steps (see below). Genes detected in fewer than three cells were excluded from the count matrices. At this step, when quality control of single cells was completed, the dataset consisted of 154,624 cells across 101 independent mice and 20 sequencing libraries (Supplementary Fig. 2d,c and Tables 5–7). For detailed sequencing statistics per library see Supplementary Tables 1–8.
Normalization and integrationAfter quality control, each library was individually normalized and scaled using SCTransform(). For all libraries, we selected the 3,000 most variable features for downstream integration. We determined a list of common integration anchors across libraries with FindIntegrationAnchors() that we used as an input for integration. To integrate all the libraries, we used the IntegrateData() function of the Seurat package to correct for any potential library batch effect. Integrated matrix was used for downstream analysis (Supplementary Fig. 2c,d).
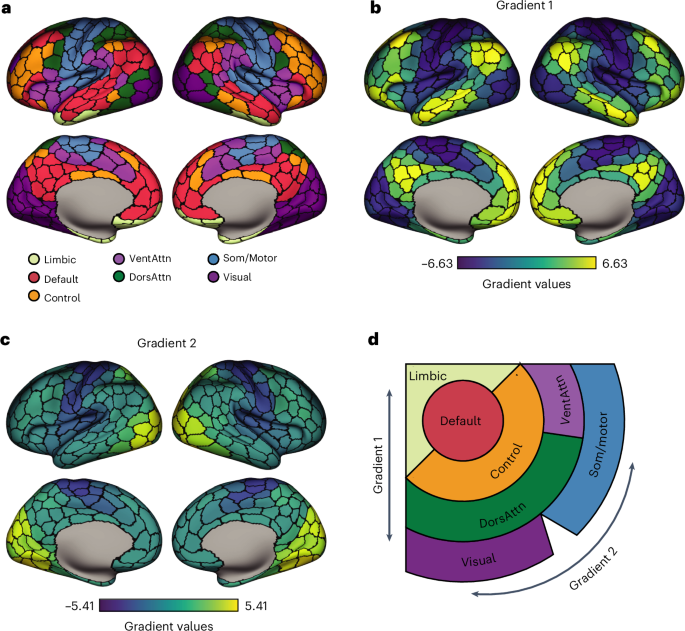
In the integrated dataset, we performed principal components analysis and found that the highest variability in the dataset was explained by the separation of CAMs from the microglial cell states, while integrated sequencing libraries did not show any batch effects in the dataset (Supplementary Fig. 2c,d). We selected 27 dimensions for dimensionality reduction by Uniform Manifold Approximation and Projection (UMAP), which we performed with the RunUMAP() function. To produce the initial UMAP as in Supplementary Fig. 3a we used the following parameters in RunUMAP(): dims = 1:27, n.neighbors = 30L, n.epochs = 200, min.dist = 0.01. To identify clusters, we first used the function FindNeighbors() (parameters for Supplementary Fig. 3a: dims = 1:20, k.param = 100, nn.method = “annoy”, annoy.metric = “cosine”) and then performed unbiased clustering by using FindClusters() (parameters for Supplementary Fig. 3a: resolution = 0.6, n.iter = 1000, n.start = 10, algorithm = 1, group.singletons = T). This led to the identification of 14 clusters, ten of which represented unique microglial/myeloid cell-state identities, three of which were merged into one homeostatic cluster for their overlapping transcriptomic signature and one of which resulted in a small low-quality cell cluster. The specific parameters used for UMAP and clustering were defined after assessment of a wide range of possible parameters, which were evaluated in light of cell-state annotations and differential expression results. We start from underclustering and we progressively increase the resolution by identifying further heterogeneity in the data, but we prevent overclustering by assessing that high resolutions lead to the definition of extra clusters that do not significantly differ in gene expression from the existing ones. Cell states were annotated by means of differential expression (FindAllMarkers() function for overall differential expression, FindMarkers() for side by side comparison) and using the AddModuleScore() function with a large number of published datasets, gene ontology categories, pathways and signatures as input3,4,7,9,14,20 (Supplementary Fig. 3b). Out of 14 clusters, nine clustered together and showed high expression of human microglial genes (P2RY12, CX3CR1, P2RY13 and so on). Three of these clusters were merged into an HM cell state for their common signature, while unique cell states were assigned to the other microglial clusters (Supplementary Fig. 3b,c, see main text and Fig. 1b–d for details). The remaining five clusters were enriched in nonmicroglial markers. Out of those, one small population clustered away from the main microglial clusters and expressed high levels of CAM markers (9,645 cells, marked by CD163, MRC1, RNASE1; Supplementary Fig. 3c,d) as previously described by Mancuso et al.14. In total, 3,396 cells clustered apart and expressed proliferation markers (high in TOP2A, MKI67, STMN1; Supplementary Fig. 3c,d). Although the dataset was previously quality controlled, a population of Low Quality/Doublets (3,517 cells) was still present. A small cluster of secretory cells was defined by sharing the ‘secretory’ signature previously identified also in Hasselman et al.7 (1,156 cells, high in AGR2, MNDA; Supplementary Fig. 3c,d). Finally, we defined a small cluster of other myeloid cells characterized by low expression of microglial homeostatic genes and expression of macrophage/monocyte markers (CD14, NEAT1, MAFB) and pro-inflammatory markers (IL1B, CCL2, CCL3) (3,078 cells; Supplementary Fig. 3c). The top differentially expressed genes from each defined cell state confirmed the unique signatures and no overclustering (top 20 marker genes per cluster are visualized in Supplementary Fig. 3c).
Microglia subsetting, re-clustering and annotationFor the final analysis of high-quality isolated human microglia, we subsetted out other myeloid and low-quality clusters (CAM, Other Myeloid, Secretory, Proliferating and Doublets/Low Quality). After trimming, 127,755 human microglia were retained for downstream analysis. Using the previously integrated dataset, we performed principal components analysis and selected 30 dimensions for dimensionality reduction by UMAP, as described above. No library-dependent batch effect was observed (Supplementary Fig. 2d). To produce the final UMAP as in Fig. 1b, we used the following parameters in RunUMAP(): dims = 1:30, n.neighbors = 30L, n.epochs = 500, min.dist = 0.05. To identify clusters, we first used the function FindNeighbors() (parameters for Fig. 1b: dims = 1:20, k.param = 100, nn.method = “annoy”, annoy.metric = “cosine”) and then performed unbiased clustering by using FindClusters() (parameters for Fig. 1b: resolution = 0.7, n.iter = 1000, n.start = 10, algorithm = 1, group.singletons = T). This led to the identification of 11 clusters, four of which were merged into an HM cell state for their overlapping transcriptomic signature (Fig. 1b,c and Supplementary Fig. 3c), and the other seven represented cell states defined by unique or transitory profiles. With the higher resolution provided by the microglia subclustering, we could identify two distinct CRM populations (CRM-1 and CRM-2) that were initially grouped together (Supplementary Fig. 3a,c) but actually represent two consecutive stages on the same phenotypic trajectory, and which can be differentially modified by Aβ pathologies and genetic backgrounds (Figs. 1b–d, 3a,b, 5d–f and 7). The specific parameters used for UMAP and clustering were defined after assessment of a wide range of possible parameters, which were evaluated in light of cell-state annotations, differential expression results and sample distributions. Cell-state annotation was performed as described for the full dataset, by means of iterative clustering, differential expression and signature scores (Fig. 1c and Supplementary Fig. 4b). We finally defined eight microglial cell states that included HM, CRM-1 and CRM-2, IRM, DAM, HLA, RM and tCRM (Fig. 1b–d). The expression profiles of the top differentially expressed genes from each defined cell state (top ten markers on heatmap in Fig. 1d, top three markers on UMAP in Supplementary Fig. 4c and all markers of differential expression statistics in Supplementary Table 8) and the signature scores calculated with AddModuleScore() (Supplementary Fig. 4a) confirmed the unique transcriptomic profiles of these clusters and that no overclustering was performed. We excluded from this dataset six mice that showed signs of infection, extremely low cell numbers and/or mice with the vast majority of cells mapping to one unique cell state. The final high-quality microglia dataset consisted of 127,755 cells from 95 independent mice and 20 sequencing libraries (Supplementary Fig. 2d,e). To extend our pseudotime analysis (Fig. 3 and Extended Data Figs. 4 and 5), we added three sequencing libraries with 10,822 single microglial cells across 11 independent mice. The total final dataset therefore includes 138,577 single cells from 106 mice and 23 sequencing libraries. For detailed sequencing statistics per library see Supplementary Table 5. For the number of sequenced cells per replicate/condition and all metadata of this study see Supplementary Tables 6 and 7.
Differential expressionDifferentially expressed genes were found by applying the FindAllMarkers() function for overall differential expression between each cluster and the rest of the dataset and FindMarkers() for side by side comparisons of two groups, both from the Seurat R package. All the reported comparisons in the manuscript were performed with the following parameters: assay = “SCT”, test.use = “wilcox”, min.pct = 0.01, logfc.threshold = 0.1. We used the Wilcoxon rank-sum test to calculate P values. We performed differential expression on the ‘SCT’ assay calculated from SCTransform(), since Pearson residuals resulting from regularized negative binomial regression effectively mitigate depth-dependent differences in differential expression, as described by Hafemeister and Satija51. We tested only genes that were detected in a minimum fraction of 1% in either of the two populations. We limited testing to genes that showed, on average, at least 0.1-fold difference (log-scale) between the two groups of cells. Only genes with their adjusted P < 0.05 (post hoc, Bonferroni correction) were considered as significant. The complete list of differentially expressed genes from all comparisons is displayed in Supplementary Tables 1, 3 and 8.
PseudotimePseudotime analysis was performed in the final human microglia dataset to infer the phenotypic transitions happening between the different microglial cell states. Unsupervised single-cell trajectory analysis was performed with Monocle 3, an algorithm that allows us to learn the sequence of gene expression changes each cell must go through as part of a dynamic biological process. We used SeuratWrappers to convert our microglial Seurat object into a Monocle object with as.cell_data_set(). We kept the UMAP embeddings previously calculated with RunUMAP() to estimate the phenotypic transitions between our annotated cell states. We ran cluster_cells() and learn_graph() (parameters used: close_loop = T, learn_graph_control = list(36an.k = 100, prune_graph = TRUE, orthogonal_proj_tip = F, minimal_branch_len = 50)) to learn the trajectory. To infer how resting microglia transition into reactive cell states, we set the roots of the trajectory with order_cells() by selecting the ten most homeostatic cells in our dataset (based on our previously defined HM signature score), to avoid limiting the selection of the root to a manually picked single cell and to account for heterogeneity of the HM cluster. Similar trials that set the origin of the trajectory to different HM cells led to comparable results, always identifying the main axes of phenotypic transitions described in Fig. 3a. To generate the trajectory displayed in Extended Data Figs. 4 and 5 including the 3-month data, we used the following parameters for learn_graph(): close_loop = F, learn_graph_control = list(rann.k = 100, prune_graph = TRUE, proj_tip = F, branch_len = 20), and we set the roots of the trajectory with order_cells() by selecting the five most homeostatic cells in the dataset.
Gene name conversion from human to mouse orthologsMouse to human orthologs tables were downloaded from Ensembl/Biomart (release 106). For the analysis reported in Extended Data Fig. 3c,d and Supplementary Table 2, mouse genes were converted to human genes with the most conservative criteria: if multiple orthologues of a mouse gene existed in human, the one with highest log2FC (fold change) in human DAM was selected. If multiple mouse genes converted to the same human gene, the one with highest log2FC in mouse DAM was selected. The overlap shown is therefore the highest that it is possible to achieve and can only be an overestimation. Note that even with the most inclusive criteria, the overlap between human and mouse DAM was very limited, and any other stricter approach (such as considering only bidirectional one-to-one orthologs) would find only lower correlations while losing information from the mouse genes.
Data exclusionWe excluded six mice from this dataset that showed signs of infection or extremely low cell numbers, and/or mice with most cells mapping to one unique cell state.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)