
Remember me
No statistical methods were used to predetermine sample sizes for the number of human samples used; however, sample sizes are the same or larger than those used in comparable studies. We consistently observed a similar distribution of tau and reported EV protein markers across all eight human donors and eight EV density fractions using mass spectrometry. As described in the Methods, three EV density fraction 8 samples (from donors A, F and E) were removed from the mass spectrometry analysis, as significantly less material was isolated from those donors. Data collection and analysis were not performed blind, as all specimens observed were from donors diagnosed with AD. Data distribution was assumed to be normal, but this was not formally tested. The statistical analyses performed for each experiment are listed in the associated figure legends. Statistical analyses were performed using GraphPad Prism.
Isolation of EVs from postmortem human brain tissueHuman tissue samples were sourced from the New York Brain Bank at Columbia University (Alzheimer’s Disease Research Center) and the University of Miami Brain Endowment Bank. Their use in this study was approved by the ethical review processes at each institution. Informed consent was obtained from the individuals’ next of kin. EVs were isolated essentially as previously described36, with minor alterations to achieve a gentler tissue dissociation suitable for fresh-frozen postmortem human brain tissue. Tissue samples had low or no time to cold (~1.5 h average) and low overall frozen postmortem intervals (~13.5 h average; Extended Data Table 1). Tissue samples were dissected and frozen from fresh using liquid nitrogen vapor to minimize freezing artifacts60. Frozen tissue (0.8–1.2 g) was transferred directly to Petri dishes to minimize thawing artifacts61 and gently sliced into pieces (1–5 mm3) using a razor blade in a few drops of prewarmed (37 °C) Hibernate A medium containing 20 U ml–1 papain (Worthington, LK003178). We avoided mincing, vortexing or homogenization, which can damage membranes62. Sliced tissue was incubated in 3.125 ml of prewarmed papain solution for 10 min at 37 °C, with three gentle tube inversions after 5 min. Papain digestion was stopped by the addition of 6.5 ml of ice-cold Hibernate A medium containing protease inhibitors (5 μg ml–1 leupeptin, 5 μg ml–1 antipain dihydrochloride, 5 μg ml–1 pepstatin A, 1 mM phenylmethanesulfonyl fluoride and 1 μM E64), and the dissociated tissue was passed slowly through a wide-bore 10-ml pipette until smooth (six to eight times). The dissociated tissue was then centrifuged at 300g for 10 min at 4 °C to pellet cells. The supernatant, containing the interstitial fluid, was passed through a 40-μm cell strainer (Corning, 352340), followed by a low-protein-binding 0.2-μm syringe filter (Corning, 431219). The filtrate was centrifuged at 2,000g for 10 min at 4 °C, and the supernatant was made to 60 ml with ice-cold PBS (Gibco, 10010-015) and further centrifuged at 10,000g for 30 min at 4 °C. The supernatant was transferred to fresh tubes and centrifuged at 100,000g for 70 min at 4 °C using a Ti45 rotor (Beckman). The 100,000g pellets were resuspended in 1 ml of ice-cold PBS and brought to 60 ml using PBS before another round of centrifugation at 100,000g for 70 min at 4 °C. The supernatants were discarded, and the pellets (containing EVs) were resuspended in 1.5 ml of 60 mM Tris-HCl (pH 7.4) containing 0.25 M sucrose and 40% Optiprep (OP) and placed in ultraclear centrifuge tubes (Beckman, 344059). Discontinuous OP gradients were prepared by layering 2 ml of 60 mM Tris-HCl (pH 7.4) containing 0.25 M sucrose and 20, 15, 13, 11, 9, 7 and 5% OP, respectively. Additional 60 mM Tris-HCl (pH 7.4) containing 0.25 M sucrose and 5% OP was then added until 1–3 mm from the top of the tube. The prepared gradients were centrifuged at 200,000g for 16 h at 4 °C in an SW41Ti rotor (Beckman) with no braking during deceleration. Eight fractions encompassing each OP concentration were collected into 30-ml thickwall polycarbonate tubes (Beckman, 355631), as described in Supplementary Fig. 1 (fraction 1 = 5% OP; fraction 2 = 7% OP; fraction 3 = 9% OP; fraction 4 = 11% OP; fraction 5 = 13% OP; fraction 6 = 15% OP; fraction 7 = 20% OP; fraction 8 = interface between the 20/40% OP layers). Fractions were diluted with 15 ml of cold PBS and centrifuged at 100,000g for 70 min at 4 °C using a 70.1Ti rotor (Beckman). The supernatants were discarded, and the pellets (containing EVs) were resuspended in 50 μl of PBS. EV suspensions were used immediately for cryo-ET and for proteinase K protection assays, or aliquoted and frozen at −80 °C for additional biochemical analyses, including before sarkosyl extraction for cryo-EM.
ImmunoblotsEV suspensions in PBS were diluted 1:1 in 2× RIPA (100 mM Tris-HCl (pH 7.4), 300 mM NaCl, 2% NP-40, 1% sodium deoxycholate and 0.2% SDS). Total protein concentration was determined using a bicinchoninic acid assay. EV suspensions were normalized to total protein concentration, prepared with 4× Laemmli sample buffer (Bio-Rad, 1610747) containing 50 mM DTT and heated at 95 °C for 5 min. Samples were resolved using 4–20% Tris-glycine extended stain-free gels (Bio-Rad). Proteins were transferred to low-fluorescence PVDF membranes (Bio-Rad) and visualized using UV light in a ChemiDoc MP imager (Bio-Rad). Membranes were subsequently blocked with 5% milk powder in Tris-buffered saline (20 mM Tris-HCl (pH 7.4) and 150 mM NaCl) containing 0.02% Tween 20 (TBST) for 1 h at 21 °C. Primary antibodies were diluted in Superblock TBS blocking buffer (Thermo Fisher Scientific) and incubated overnight with the membranes. Membranes were then washed twice in TBST and twice in TBST containing 5% milk powder, with each wash lasting 10 min. Secondary horseradish peroxidase (HRP)-conjugated antibodies were diluted in TBST containing 5% milk powder and incubated with the membranes for 1 h at 21 °C. Membranes were then washed four times in TBST for 10 min each wash, and labeled proteins were visualized using Bio-Rad Clarity Max chemiluminescent substrate in a ChemiDoc MP imager. The total protein signal visualized using UV light was used to normalize the labeled protein signal using the Image Lab software package, version 6.1, build 7 (Bio-Rad). The following primary antibodies were used: annexin A2 (Cell Signaling, D11G2; 1:1,000), flotillin 1 (BD Transduction, 610821; 1:1,000), CD81 (Cell Signaling, D3N2D; 1:1,000), LAMP1 (Abcam, ab62562; 1:250), LAMP2 (Santa Cruz, sc-18822, clone H4B4; 1:500), VDAC (Cell Signaling, 4866; 1:1,000), lamin A/C (Santa Cruz, sc-376248; 1:1,000), Tau13 (BioVision, 3453-100; 1:1,000), HT7 (Invitrogen, MN1000; 1:1,000), TauC (Dako, A0024; 1:5,000), PHF1 (mouse monoclonal; gift from P. Davies; 1:250), EEA1 (BD Biosciences, 610457; 1:250), β-actin–FITC (Sigma-Aldrich, F3022; 1:1,000), MC1 (gift from P. Davies; 1:250), phospho-tau-T217 (Thermo Fisher Scientific, PA5-37639; 1:250) and phospho-tau-S422 (Thermo Fisher Scientific, 44-764G; 1:250).
Dot blotsSarkosyl-insoluble extracts from pooled EV fractions 4–6 as well as sarkosyl-insoluble extracts from total brain homogenates were transferred onto nitrocellulose membranes (0.2-µm pore size; Bio-Rad, 162-0112) using the 96-well Minifold I Dot-Blot System (Whatmann). The membranes were incubated in a blocking buffer containing PBS supplemented with 0.1% Tween 20 (PBST) and 1% bovine serum albumin (BSA) for 30 min at 21 °C, followed by incubation in blocking buffer containing the antibody MC1 (1:250) overnight at 4 °C. The membranes were washed three times in PBST for 10 min each wash and incubated in HRP-conjugated secondary antibodies (Bio-Rad; 1:5,000) for 1 h at 21 °C. After washing three times in PBST for 10 min each wash, the membranes were incubated with Enhanced Chemiluminescence Prime reagents (Amersham) for 1 min at 21 °C and imaged using a ChemiDoc MP (Bio-Rad). The membranes were washed three times in PBST for 10 min each wash and incubated in blocking buffer for 30 min at 21 °C before incubation in blocking buffer containing the antibody TauC (Dako, A0024; 1:5,000) for 1 h at 21 °C. After washing the membrane three times in PBST for 10 min each wash, the membranes were incubated in DyLight 800-conjugated secondary antibodies (Cell Signaling; 1:2,500) for 1 h at 21 °C. The membranes were washed three times (10 min each) in PBST and imaged using a ChemiDoc MP (Bio-Rad).
Proteomic sample preparation and liquid chromatography–tandem mass spectrometry analysisReduction, alkylation, S-TRAP clean-up and protease digestionEV fractions were resuspended in TBS (20 mM Tris-HCl (pH 7.4) and 150 mM NaCl) containing 5% SDS, and proteins were digested using S-TRAP Micro spin columns, according to the manufacturer’s protocol (Profiti). Approximately 50 μl of starting material was used for each experiment, corresponding to ~50 μg of total protein. Samples were reduced by the addition of 20 mM DTT and incubated for 30 min at 21 °C. Samples were alkylated by the addition of 40 mM iodoacetamide for 30 min at 21 °C protected from light. Phosphoric acid was then added to each sample to 1.2%, followed by vortexing. Samples were diluted 6.6-fold in S-TRAP Bind and Wash buffer composed of 90% methanol containing 100 mM tetraethylammonium bromide (TEAB; pH 8) and loaded into an S-TRAP Micro spin column by centrifugation at 4,000g at 21 °C until all of the solution passed through the column. Each spin column was subsequently washed with 150 μl of S-TRAP Bind and Wash buffer and centrifuged at 4,000g for 30 s at 21 °C. The flow-through was discarded. This wash step was repeated three more times for a total of four washes. Before digestion, spin columns were transferred to clean collection tubes. Porcine trypsin (Promega) in 50 mM TEAB (pH 8) was added to each spin column to a final enzyme:substrate ratio of 1:10, followed by incubation for 2 h at 47 °C without agitation. Digested peptides were eluted from the spin columns by the addition of 40 μl of S-TRAP Elution Buffer A composed of 50 mM TEAB (pH 8) and centrifugation at 4,000g for 30 s at 21 °C. A second elution was performed by the addition of equal volumes of S-TRAP Elution Buffer B composed of 0.5% trifluoroacetic acid in water and S-TRAP Elution Buffer C composed of 0.5% trifluoroacetic acid in 50% acetonitrile. The eluates were combined, dried using a speedvac and resuspended in 0.1% formic acid (FA) for subsequent mass spectrometry analysis.
Evosep/timsTOF liquid chromatography–tandem mass spectrometryA high-throughput liquid chromatography–tandem mass spectrometry workflow was implemented as described in Jones et al.63. EvoTips (Evosep) were activated by soaking in propanol and washed with 20 μl of 0.1% FA in acetonitrile (solvent B) with centrifugation at 700g for 60 s. Tips were conditioned by soaking in propanol until the C18 material appeared pale white, centrifuged at 700g for 60 s and equilibrated with 20 μl of 0.1% FA in water (solvent A). To prevent drying out, samples were loaded into tips while equilibrating in solvent A. Peptides were bound to the C18 material by centrifugation at 700g for 60 s. Tips were subsequently washed with 20 μl of solvent A and centrifuged at 700g for 60 s. Following this, 100 μl of solvent A was added to the tips, which were then centrifuged at 700g for 10 s. Liquid chromatography–tandem mass spectrometry analysis was performed immediately. Peptides were analyzed using an Evosep One (Evosep) coupled to a timsTOF Pro mass spectrometer (Bruker), with separation of peptides on a C18 column (150 μm × 80 mm) packed with 1.5-μm beads (PepSep). A gradient length of 21 min at a flow rate of 1 μl min–1 was used. The timsTOF Pro mass spectrometer was operated in a parallel accumulation, serial fragmentation (PASEF) mode. Trapped ion mobility spectrometry (TIMS) ion accumulation and ramp times were set to 100 ms, and mass spectra were recorded from m/z 100 to 1,700. diaPASEF used eight diaPASEF scans per TIMS–mass spectrometry scan, giving a duty cycle of ~1 s. The ion mobility range was set to 0.85–1.3 Vs cm–2. Each mass window isolated was m/z 25 wide, ranging from m/z 475 to 1,000 and with an ion mobility-dependent collision energy that increased linearly from 27 eV to 45 eV between 0.85 and 1.3 Vs cm–2.
Data searchingData-independent acquisition data were searched using default settings in DIA-NN software (version 1.8). Methionine oxidation was set as a variable modification, and cysteine carbamidomethylation was set as a fixed modification. The search allowed for one missed trypsin cleavage site. Samples were searched against the Homo sapiens UniProt proteome (retrieved in February 2021) and quantified by label-free quantitation (LFQ).
Mass spectrometry data analysisRaw protein intensities were analyzed in R 4.2.1 (ref. 64), with all code documented within R Markdown65 at https://github.com/duff-lab-team/AD-EV-characterisation. Reproducibility and package versioning is provided through Singularity/Docker56. The following pipeline was used for both protein and peptide LFQ intensities. Intensity data were imported and handled using the Differential Enrichment analysis of Proteomics data package66. A total of 6,105 unique proteins were identified across all the fraction groups (Supplementary Fig. 2a). Three fraction 8 samples (A, F and E) were excluded from the analysis because significantly less material was isolated from those donors. After filtering (described in the flowchart below), 6,054 proteins were retained for the imputation pipeline.
Missing data (Supplementary Fig. 2b,c) were handled using a hybrid missing-not-at-random (MNAR) and missing-at-random (MAR) imputation strategy designed to distinguish between proteins that were completely absent from a particular fraction (likely MNAR) and proteins with values missing only from a small number of donors or fractions (likely MAR). This hybrid strategy maximizes the inclusion of differentially expressed proteins (DEPs) across the fractions and avoids exclusion of biologically relevant protein identities (for example, proteins expressed selectively in one or two fraction groups)67. This strategy led to a 43% increase in the number of DEPs compared to if the analysis was restricted to proteins without missing values in any of the fractions.
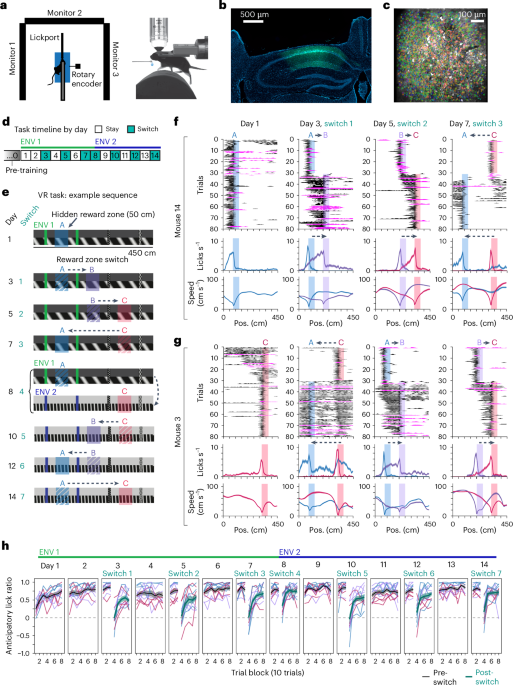
Design and implementation of the hybrid imputation strategyTo demonstrate the effectiveness of the hybrid imputation strategy and aid in the selection of MAR versus MNAR missing values, a simulated dataset was produced from a matrix of 6,105 complete case log2 (intensity values) based on the mean and standard deviation of proteins in the real dataset67. To model the characteristics of the real dataset, 3,000 DEPs and missing values of both MAR and MNAR type were manually assigned to the simulation. The relationship between true missing values across different mean intensities with MAR alone (Supplementary Fig. 3a) versus MNAR + MAR (Supplementary Fig. 3b) was investigated to define a cutoff for MNAR missing values. In the simulation, the MNAR cut-off was set at the bottom of the steep decline in true missing values (log2 intensity value of 12); Supplementary Fig. 3b). This maximized the number of true DEPs captured, the accuracy of their detection and the average adjusted F-test P value of an analysis of variance across all three fraction groups (Supplementary Fig. 4a). Imputation of the simulated dataset identified 92.97% of the true DEPs with similar accuracy as using the complete cases, while only 69.07% and 24.03% of true DEPs were captured in the unimputed and complete cases (Supplementary Fig. 4b,c). To apply this strategy to the real dataset, MNAR log2 (intensity cutoff) values were manually determined for each fraction using the simulation curve as a guide (Supplementary Fig. 3c): fraction 1, 14.9; fraction 2, 14.7; fraction 3, 14.9; fraction 4, 14.3; fraction 5, 14.6; fraction 6, 14.5; fraction 7, 15.3; fraction 8, 15.2. In the real dataset, 827 more proteins were detected as DEPs following imputation (Supplementary Fig. 4d). Data were normalized using a variance stabilizing transformation, and principal component analysis was performed and visualized using the R package ggplot2 (ref. 68). The 68% confidence interval was used to draw ellipses surrounding the points for each donor (Fig. 1b).
Differential expression and Gene Ontology pathway analysisDifferential expression analysis was performed using moderated F- and t-statistics from the R package limma69,70, with visualization aided by the R packages volcano3D60 and ComplexHeatmap71. Pairwise contrasts were tested across the three fraction groups using the Benjamini and Hochberg multiple testing correction factor. Proteins were deemed significant at adjusted P values of <0.05, without a log fold change cutoff. Gene Ontology analysis (Fig. 1c) was performed using the ssGSEA method from the GSVA R package72. After the ssGSEA transformation, the same limma testing procedure was applied to the new matrix, with a more stringent threshold of adjusted P < 5 × 10−4. To reduce multiple testing burden, data were filtered such that only gene sets with between 5 and 50 genes detected were used. Gene sets with more than 25% overlapping genes and gene sets with directionality modifiers as ‘up/downregulation of’ and so on were discarded. Relevant terms with the highest F-test P values across the fraction groups were selected for display in Fig. 1c.
HEK cell model of tau assemblyHEK293T cells (ATCC, CRL-3216) were transduced with lentivirus expressing 1N4R tau with the P301S mutation fused to enhanced YFP at the C terminus. A clonal population was obtained by fluorescence-activated cell sorting and was seeded with PHFs isolated from human AD brain tissue (Extended Data Table 1). The seeded population was again sorted, and clones were selected that constitutively propagated assembled tau, as visualized by the presence of YFP+ cytoplasmic puncta. Cells were cultured in complete medium (DMEM; Gibco, 41966029) supplemented with 10% fetal bovine serum (Gibco, 10082-147) and 1% penicillin/streptomycin (Gibco, 15140-122) in flasks coated with 20 μg ml–1 poly-d-lysine (Sigma, P6403). Confocal images were acquired using a Leica Stellaris 8 STED microscope with a ×100 oil objective (Extended Data Fig. 4a).
Isolation of EVs from HEK cell mediumWhen expanding cells for EV collection, the cells were plated as described above in complete medium and grown to 50% confluency. Serum-containing medium was removed, cells were washed gently in warmed PBS, and the medium was replaced with serum-free Advanced DMEM (Gibco, 12491015) supplemented with 2 mM l-glutamine (Gibco, A2916801) and 1% penicillin/streptomycin (Gibco, 15140-122). The medium was pipetted from cells at 100% confluency and centrifuged for 10 min at 2,000g to pellet cells and large debris. Medium supernatants were stored at −80 °C. For each EV isolation, approximately 360 ml of collected medium was centrifuged at 150,000g at 4 °C for 16 h to generate a crude EV pellet, which was resuspended in 30% OP and overlayed with 20% and 10% OP layers. Gradients were centrifuged as per the human EVs. All floated material (including the 30–20% interface) was pooled, diluted 1:15 in PBS (Gibco, 10010-015) and centrifuged at 150,000g for 3 h to collect an EV pellet.
NTA particle analysisAt isolation, fresh EVs were partitioned into 3-μl aliquots for particle counting and frozen at −80 °C. Samples were thawed only once before NTA analysis (ZetaView PMX-420 Quatt, Particle Metrix) and immediately diluted in particle-free PBS to the optimal reading range of the instrument (100–150 particles). Samples were diluted in PBS (between 1:100,000 and 1:5,000) and analyzed at 22 °C in scatter mode at the following settings: sensitivity = 80, shutter = 100, frame rate = 30, trace length = 15, bin size = 5 nm and positions per single reading = 11. Code and R Markdown sheets describing the data analysis are deposited at https://github.com/duff-lab-team/AD-EV-characterisation. In brief, data files for each EV sample were aggregated into a single data object and normalized by multiplying the concentration at each size bin by the corresponding dilution factor for that sample. To obtain the number of EVs per milligram of brain tissue (Extended Data Fig. 2e), a scaling factor was applied to normalize each sample to the amount of tissue isolated, and means of three to five technical replicates were calculated. For all other NTA figures, particle counts were used for estimation of the mode, and summing of counts was used to aggregate technical replicates.
Seeded tau assembly in Tau RD P301S FRET Biosensor cellsTau RD P301S FRET Biosensor cells (ATCC, CRL-3275) were plated at 30,000 cells per well in 96-well flat-bottomed plates in complete medium (as described above). The cells were allowed to settle undisturbed for 15 min at room temperature before being moved to the incubator and grown at 37 °C with 5% CO2. The following day, seed mixes were made in 20 μl per well volumes containing 15 μl of protein dilution (3 μg of EVs in PBS diluted in a final volume of 15 μl of Opti-MEM (31985062, Gibco)) and 5 μl of Lipofectamine dilution (1 μl of Lipofectamine 2000 plus 4 μl of Opti-MEM). Seeding reactions were incubated for 30 min in the cell culture incubator before being added to cells 24 h after plating (cells were ~65–70% confluent at the time of seeding). The seeded cells were maintained undisturbed in the incubator for 48 h before collection for FRET measurements.
At the time of collection, the cell medium was removed from each well using a vacuum manifold for 96-well plates. Trypsin-EDTA (50 μl) was added to each well, and the plate was incubated for 2–3 min at 37 °C. Serum-containing medium (150 μl) was added to each well, and cells were triturated ten times and moved into wells of a fresh v-bottomed 96-well plate. Cells were pelleted at 600g for 5 min and resuspended in 150 μl of cold 4% paraformaldehyde in PBS by triturating ten times. Cells were left to fix for 10 min and then repelleted at 600g for 5 min. The cells were resuspended in 200 μl of PBS containing 1 mM EDTA. Flow cytometry analysis of FRET signals was performed using a BD LSR Fortessa flow cytometer equipped with a high-throughput sampler using BV510 and BV421 BD filter sets, as previously described73. Samples were run on the ‘low’ flow setting (<1,000 events per s), and 50,000 events were captured per well, with three technical replicates for each condition. Integrated FRET densities (percent FRET-positive cells per gated cell population × median fluorescence intensity of the BV510 signal) were obtained using FCS Express 7 Research software (De Novo Software).
Seeded tau assembly in PS19 miceAll experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Columbia University. Three 6-month-old male PS19 mice were injected with pooled AD EVs (fractions 1–8) in the left hemisphere and pooled control EVs (fractions 1–8) in the right hemisphere. Mice were immobilized in a stereotaxic frame (David Kopf Instruments) under continuous isofluorane administration, and stereotaxic injections were made under aseptic conditions using a Hamilton syringe into the hippocampal hilus (anterior–posterior, –2.5 mm; medial–lateral, ±2 mm; dorsal–ventral, −1.8 mm). Each injection contained 10 μg of total EV protein in a volume of 1.5 μl. All animals were carefully monitored and administered analgesics both during and after surgery. Mice were killed 2 months after injection and perfused intracardially with PBS and 10% formalin. Brains were postfixed overnight in 10% formalin and cryoprotected in 30% sucrose for 1 week before TissueTek embedding. Thirty-micron-thick coronal sections spanning the anterior–posterior axis of the hippocampus (bregma –1.5 to 3) were prepared using a Leica cryostat. Free-floating sections were immunolabeled for AT8 using the Mouse-on Mouse (MOM) Elite peroxidase immunodetection kit (Vector Laboratories, PK-2200). Briefly, endogenous peroxidases were quenched in hydrogen peroxide buffer (PBS + 10% methanol + 3% H2O2) for 15 min at 21 °C. Tissue sections were washed twice in PBS, blocked using the mouse IgG blocking reagent for 1.5 h at 21 °C and washed again three time with PBS. AT8-biotin (Thermo Fisher Scientific, MN1020B) primary antibody was incubated with the sections at a dilution of 1:500 in the MOM diluent overnight at 4 °C. Sections were washed five times in PBS, incubated for 30 min at 21 °C in ABC working solution (Vectastain Elite HRP kit; Vector laboratories, PK-6100) and washed again three times in PBS. 3,3′-Diaminobenzidine (DAB) working solution (DAB Peroxidase Substrate kit; Vector Laboratories, SK-4100) was added to the sections until the color was fully developed (under constant dissection microscope monitoring). Sections were immediately washed twice in PBS to stop the reaction, mounted onto glass coverslips and dehydrated successively in 70% ethanol, 95% ethanol, 100% ethanol and 100% xylene. Coverslips were mounted using Permount (DPX mounting medium) and dried overnight. DAB labeling was assessed every fifth slice (ten slices per animal) at ×20 magnification on a Zeiss Axio Scan microscope. AT8 immunolabeling intensity was quantified from ipsilateral (AD EV-injected) and contralateral (control EV-injected) brain regions (dentate gyrus, CA2/CA3) using ImageJ.
Isolation of EVs spiked with sarkosyl-insoluble material from AD brainSarkosyl-insoluble material enriched with tau PHFs was extracted from 1 g of gray matter from the frontal cortex of an individual who had AD, based on Guo et al.12. Gray matter was homogenized in 9 volumes of ice-cold extraction buffer containing 10 mM Tris-HCl (pH 7.4), 0.8 M NaCl, 10% sucrose, 1 mM EDTA, 0.1% sarkosyl and protease/phosphatase inhibitor cocktail (Pierce) using a FastPrep-24 beat-beating homogenizer (MP Biomedicals). The homogenate was centrifuged at 9,296g for 10 min at 4 °C using a Ti45 rotor (Beckman). The supernatant was filtered through a 70-μm pore-size cell strainer and retained on ice. The pellet was resuspended in 4.5 volumes of ice-cold extraction buffer and recentrifuged at 9,296g for 10 min at 4 °C using a Ti45 rotor (Beckman). The supernatant was filtered through a 70-μm pore-size cell strainer and combined with the first supernatant. The combined filtered supernatant was brought to a final concentration of 1% sarkosyl using 25% sarkosyl in water and incubated for 1 h at 21 °C with stirring at 100 rpm. Following incubation, the supernatant was centrifuged at 139,662g for 75 min at 4 °C using a Ti45 rotor (Beckman). The floating lipid layer and supernatant were discarded, and the centrifuge tube was washed twice with 6 ml of PBS, followed by one wash with 2 ml of PBS, taking care not to disturb the pellet. The pellet was transferred to a Ti70 centrifuge tube (Beckman) in 3 ml of PBS and vortexed. The tube was topped up to 23 ml with PBS and centrifuged at 259,939g for 30 min at 4 °C using a Ti70 rotor (Beckman). The floating lipid layer and supernatant were discarded, and the pellet was transferred to a 2-ml tube in 50 μl of PBS per g of initial gray matter. The tube was incubated with rocking at 25 rpm for 16 h at 21 °C to soften the pellet before being resuspended by sonication using a Hielscher S26D11X10 Vial-Tweeter-Sonotrode with 0.5-s pulses at 100% amplitude for a total accumulated power of 200 W. The resuspended pellet was transferred to a Ti70 centrifuge tube (Beckman) and centrifuged at 116,982g for 40 min at 4 °C using a Ti70 rotor (Beckman). The supernatant was discarded, and the pellet was transferred to a 2-ml tube in 50 μl of PBS per g of initial gray matter and resuspended by sonication using a Hielscher S26D11X10 Vial-Tweeter-Sonotrode in 0.5-s pulses at 100% amplitude for a total accumulated power of 200 W. The resuspended pellet was centrifuged at 10,000g for 30 min at 4 °C, and the supernatant, containing extracted tau PHFs, was retained. EVs from 0.3 g of control and AD frontal cortex were isolated as described above but were floated in a simplified OP gradient consisting of 3-ml step gradients of 40, 30, 20, 10 and 0% OP. Immediately before papain digestion, 15 μl of tau PHFs were added to the control EVs to achieve the same amount of tau PHFs that would have been isolated from 0.3 g of AD tissue.
Tau peptide mappingIndividual tau peptide LFQ intensities were imputed and normalized using the same strategy as the protein dataset and were presented as log2 values. Peptides were arranged from N terminus to C terminus, amino acids 1–441. Near-duplicate peptides, corresponding to missed tryptic cleavages and post-translationally modified peptides, were summed to represent the total abundance of sequenced peptides at each amino acid site. Peptides were treated as individual peptides if they were more than 50% unique from a neighboring peptide. A resultant 17 peptides between amino acids 6 and 406 were used in the analysis.
Sarkosyl extraction of EVsEV suspensions were pelleted by centrifugation at 100,000g for 70 min at 4 °C using a 70.1Ti rotor (Beckman) and resuspended in 20 volumes (wt/vol) of extraction buffer containing 10 mM Tris-HCl (pH 7.5), 0.8 M NaCl, 10% sucrose, 1 mM EGTA and 1% sarkosyl. The resuspended EVs were incubated with rotation for 1 h at 21 °C, followed by centrifugation at 100,000g for 70 min at 4 °C. For single-particle cryo-EM, the sarkosyl-insoluble material was further washed by two rounds of resuspension, followed by repelleting by centrifugation at 100,000g for 70 min at 4 °C, first using 25 μl of extraction buffer per g of initial tissue used and the same volume of 20 mM Tris-HCl (pH 7.4) and 100 mM NaCl. The final sarkosyl-insoluble pellets were resuspended in 25 μl of 20 mM Tris-HCl (pH 7.4) and 100 mM NaCl per g of initial tissue used. For Fig. 2d, each EV fraction was resuspended in 30 μl of PBS and pooled into three groups: fractions 1–3, 4–6 and 7 and 8. Equal volumes of each fraction group were sarkosyl extracted as described above but underwent a 10-min clearing spin at 10,000g after the sarkosyl incubation. An additional wash in 1% sarkosyl buffer was also performed to clear the pellets of lipids. Final sarkosyl-insoluble pellets were sonicated at 65% amplitude for 2 min in 1× Laemmli sample buffer (Bio-Rad, 1610747) containing 50 mM DTT and heated at 95 °C for 5 min before electrophoresis.
Proteinase K protection assayDirectly after centrifugation, each freshly prepared EV pellet was resuspended in 30 μl of PBS. Fractions 1–3, 4–6 and 7 and 8 were pooled and brought to 1 μg µl–1 total protein in PBS. Protease reactions were set up in a total volume of 15 μl with 1.5 μl of the pooled EV sample (1.5 μg of total protein) and 1 μl of 15 ng μl–1 proteinase K stock (Thermo Fisher Scientific, EO0491) to maintain a ratio of 10 ng of proteinase K:1 μg of total protein. For EV permeabilization, 1 μl of 1% Triton X-100 stock was added. Reactions were incubated at 37 °C for 30 min and stopped by the addition of 15 μl of 2× Laemmli sample buffer + 2× HALT protease and phosphatase solution (Thermo Fisher Scientific, 78440) and heating for 5 min at 95 °C. The reactions were resolved by SDS–PAGE and immunoblotted with antibody TauC at a dilution of 1:5,000.
EV fractionation and carbonate membrane purificationEVs were isolated as described above, however, before flotation in the OP gradient, the crude EV pellet was resuspended in 1 ml of PBS containing 0.1 M DTT and incubated at 21 °C for 30 min with end-over-end rotation. DTT-treated EVs were pelleted at 100,000g for 70 min at 4 °C in a TLA100 rotor (Beckman). The supernatant, containing DTT-labile EV surface proteins, was concentrated to under 100 μl in 0.5-ml, 3-kDa molecular weight cutoff centrifugal concentrators (Millipore, UFC500396; surface sample in Fig. 4c). The pelleted DTT-treated EVs were resuspended in 5 ml of 40% OP, layered beneath a step gradient of 2.5 ml each of 20, 10 and 0% OP in Buffer B containing 0.1 M DTT and centrifuged at 200,000g for 16 h at 4 °C. The top 11 ml and the bottom 2 ml of the gradient were transferred to separate 60-ml 45Ti ultracentrifuge tubes (Beckman), topped up with PBS and centrifuged at 100,000g for 70 min at 4 °C. The pellet from the 2-ml fraction from the bottom of the gradient was resuspended in 100 μl of TBS (pellet sample in Fig. 4c). The pellet from the 11-ml fraction from the top of the gradient was resuspended in 1.1 ml of hypotonic buffer consisting of 5 mM potassium phosphate, passed through a 26-gauge needle 20 times to lyse EVs and extract luminal proteins and centrifuged at 150,000g for 3 h at 4 °C in a TLA100 rotor. The supernatant, containing luminal proteins, was concentrated to under 100 μl in 0.5-ml 3-kDa molecular weight cutoff centrifugal concentrators (luminal sample in Fig. 4c). The pellet, containing EV membranes, was resuspended in 5 ml of 40% OP and layered beneath another step gradient as described before but in the absence of DTT. The gradient was centrifuged and fractionated as described before, except that the pellet collected from the 11-ml fraction from the top of the gradient was resuspended in 1.1 ml of 0.1 M sodium carbonate buffer (pH 11) and incubated for 30 min at 21 °C to extract all but integral membrane proteins from the EV membranes. The carbonate-stripped membranes were collected by centrifugation at 150,000g for 3 h at 4 °C in a TLA100 rotor. The pellet was subjected to a final OP step gradient without DTT, and the 11-ml fraction from the top of the gradient was pelleted at 150,000g for 3 h. The final pellet was resuspended in 100 μl of TBS with protease inhibitors (membrane sample in Fig. 4c). Equal proportions of each compartment subfraction were resolved by SDS–PAGE and immunoblotted with antibody TauC at a dilution of 1:5,000.
Cryo-EM assessment of EVs associated with tau filamentsEVs associated and not associated with tau filaments were manually identified in medium-magnification maps of entire EM grids using cryo-EM. EVs associated with tau filaments were verified using cryo-ET. To estimate the proportion of EVs associated with tau filaments in pooled EV fractions 4–6, average counts from two grid squares from two different EM grids were taken.
Cryo-ETPooled EV fractions 4–6 isolated from 0.8 g of tissue were diluted 1:5 in PBS containing a 1:3 dilution of 10-nm gold-conjugated BSA (BBI solutions), applied to glow-discharged 2/2-μm holey carbon-coated 200-mesh gold grids (Quantifoil) and plunge-frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). Tilt series were acquired using a 300-keV Titan Krios microscope (Thermo Fisher Scientific) equipped with a K3 detector (Gatan) and a GIF-quantum energy filter (Gatan) operated with a slit width of 20 eV. Tilt series were acquired at a pixel size of 2.13 Å or 1.89 Å at a target defocus of −3 μm to −6 μm. A dose-symmetric acquisition scheme was implemented in Serial EM over a tilt range of ±60° at 3° increments74,75. Each tilt received a dose of 3.17 e− Å–2 fractionated over eight movie frames, resulting in a total dose of 130 e− Å–2 per tilt series.
Tomogram processing and analysesGain correction, motion correction and contrast transfer function (CTF) estimation were performed in Warp76. Tilt series alignment using the 10-nm gold-conjugated BSA as fiducials was performed in IMOD77. Tomograms were reconstructed in Fourier space at a pixel size of 8 Å or 12 Å, filtered using Wiener-like deconvolution and denoised using the Noise2Noise machine learning principle78, all in Warp. For filament and EV measurements, IMOD was used to perform gain correction, motion correction, contrast function estimation and tomogram reconstruction using weighted backprojection at a pixel size of 8.51 Å or 7.46 Å. Filament widths and helical crossover distances were measured manually. Three-dimensional representation and segmentation were performed using napari-tomoslice (https://github.com/alisterburt/napari-tomoslice) in Napari79.
Subtomogram averagingSubtomogram averaging was performed based on Burt et al.80, with adaptations for helical reconstruction. Tomograms were reconstructed in Warp at a pixel size of 16 Å to aid in the visualization of tau filaments. Initial processing steps were performed in Dynamo81. Filaments were manually picked using the filament with torsion model, and 17,323 subtomograms were extracted using a box size of 64 pixels at an intersubtomogram distance of 16 Å. An initial reference was created by aligning and averaging a random subset of 250 subtomograms and was subsequently used to align and average all subtomograms. Subtomograms within 16 Å of one another were reduced to a single subtomogram using the remove_duplicates.m script (https://github.com/teamtomo/teamtomo.github.io/tree/master/walkthroughs/EMPIAR-10164/scripts). The metadata table for the remaining 6,133 subtomograms was converted into the Self-defining Text Archiving and Retrieval (STAR) format using the dynamo2m package (https://github.com/alisterburt/dynamo2m). The filament number (Helical Tube ID) for each subtomogram, specified in column 21 of the Dynamo metadata table, was added to the STAR file using the Starparser package (https://github.com/sami-chaaban/starparser). The subtomograms were then re-extracted in Warp at a pixel size of 6 Å and a box size of 204 pixels. Subsequent processing was performed using helical reconstruction methods in RELION-3.1 (ref. 82). An initial model was generated by averaging a random subset of 500 subtomograms using relion_reconstruct. Three-dimensional autorefinement followed by 3D classification were then performed without masking or applying symmetry. This yielded reconstructions corresponding to filament types resembling PHFs and SFs. The helical crossover distances were measured from the reconstructions and used to calculate the helical twists for a helical rise of 4.7 Å. Helical symmetry was then applied in all subsequent steps. Three-dimensional classification was repeated to obtain final subsets of subtomograms for PHF and SF types, followed by unmasked and masked 3D autorefinements. The subtomograms were then re-extracted in Warp at pixel sizes of 3.1 Å (PHFs) and 3 Å (SFs), with respective box sizes of 165 pixels and 170 pixels. Three-dimensional autorefinement was repeated, with C2 symmetry imposed for the PHF type. The final reconstructions were sharpened using the standard postprocessing procedures in RELION-3.1, and overall resolutions were estimated at a Fourier shell correlation of 0.143 between the two independently refined half-maps using phase randomization to correct for convolution effects of a generous, soft-edged solvent mask. Helical symmetry was imposed using the RELION Helix Toolbox. Rigid-body fitting of models of PHFs from EVs and SFs from primary age-related tauopathy (Protein Data Bank (PDB) ID 7NRS) was performed in ChimeraX83.
Single-particle cryo-EMSarkosyl-insoluble extracts were centrifuged at 10,000g for 10 min at 4 °C. The supernatants were retained and centrifuged at 100,000g for 1 h at 4 °C. The pellets were resuspended in 30 μl of 20 mM Tris-HCl (pH 7.4) containing 100 mM NaCl and centrifuged at 3,000g for 30 s at 21 °C. The supernatants were retained and applied to glow-discharged 1.2/1.3-μm holey carbon-coated 300-mesh gold grids (Quantifoil) and plunge-frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). Images for the first EV dataset were acquired using a 300-keV Titan Krios microscope (Thermo Fisher Scientific) equipped with a K2 detector (Gatan) at the European Synchrotron Radiation Facility (ESRF)84. Images for the second EV dataset and the cell dataset were acquired using a 300-keV Titan Krios microscope (Thermo Fisher Scientific) equipped with a K3 detector (Gatan) at the MRC Laboratory of Molecular Biology. GIF-quantum energy filters (Gatan) operated at slit widths of 20 eV and aberration-free image shift within the EPU software (Thermo Fisher Scientific) were used during image acquisition. Further details are provided in Extended Data Table 2.
Helical reconstructionMovie frames were gain corrected, aligned, dose weighted and summed using the motion correction program in RELION-4.0 (ref. 85). The motion-corrected micrographs were used to estimate the CTF using CTFFIND-4.1 (ref. 86). All subsequent image processing was performed using helical reconstruction methods in RELION-4.0 (ref. 87). The filaments were picked manually, and reference-free 2D classification was performed to remove suboptimal segments. Initial 3D reference models were generated de novo by producing sinograms from 2D class averages as previously described88. Three-dimensional autorefinements with optimization of the helical twist were performed, followed by two cycles of Bayesian polishing and CTF refinement85. Three-dimensional classification was used to further remove suboptimal segments, after which the 3D autorefinement, Bayesian polishing and CTF refinement procedures were repeated. The final reconstructions were sharpened using the standard postprocessing procedures in RELION-4.0, and overall resolutions were estimated at a Fourier shell correlation of 0.143 between the two independently refined half-maps using phase randomization to correct for convolution effects of a generous, soft-edged solvent mask89. Local resolution estimates were obtained using the same phase randomization procedure but with a soft spherical mask that was moved over the entire map. Helical symmetry was imposed using the RELION Helix Toolbox. Further details are provided in Extended Data Table 2.
Atomic model building and refinementThe published atomic model of PHFs from the frontotemporal cortex of an individual with sporadic AD (PDB: 6HRE) was fitted to the final reconstructions using ChimeraX. The fitted models were subsequently refined in real-space using COOT
Comments (0)