
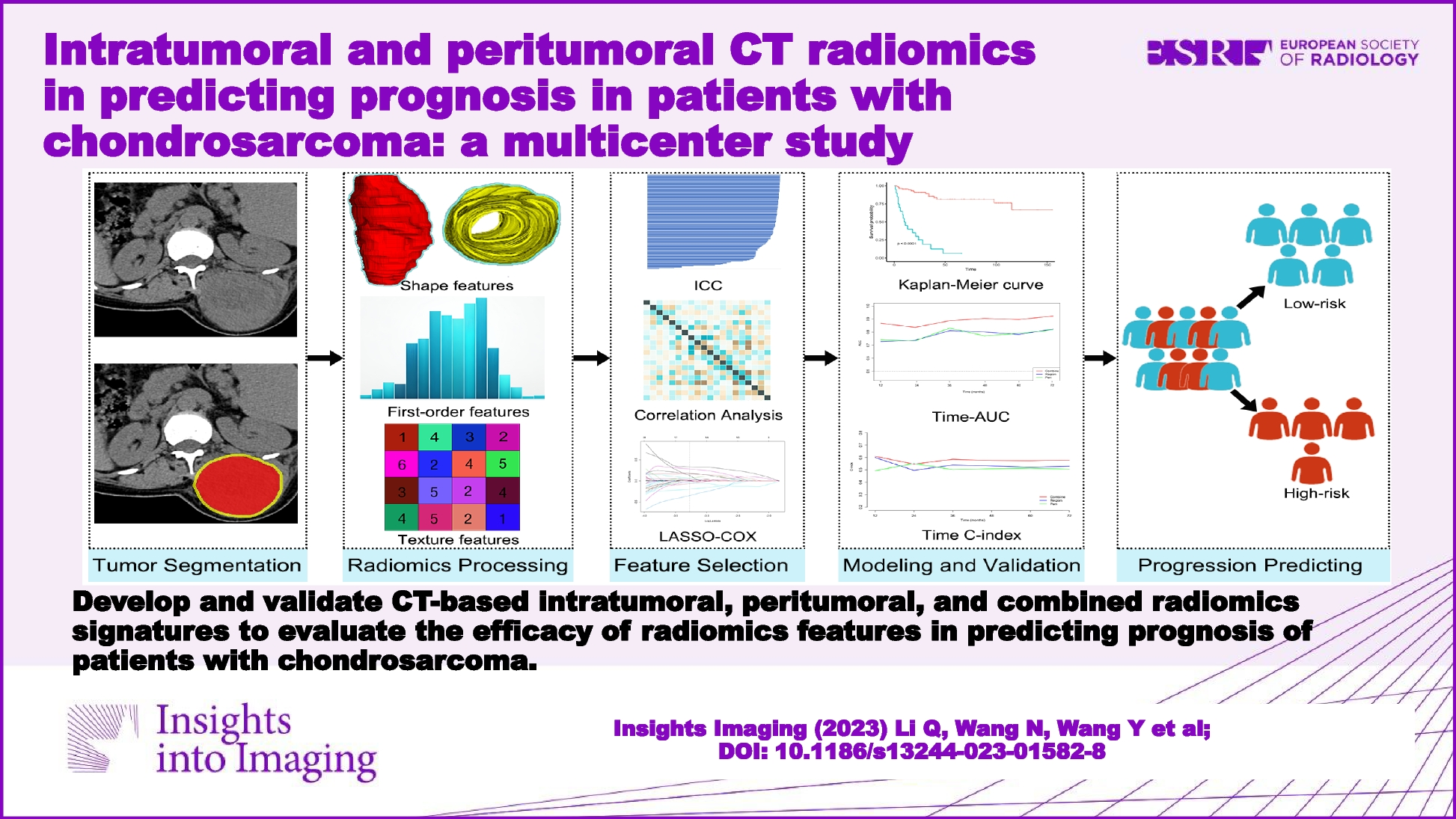
Remember me





The ethics committee of the hospital granted approval for this retrospective study, and the need for written informed consent was waived. The Cancer Genome Atlas Kidney Clear Cell Carcinoma dataset comprising 237 ccRCC patients was obtained from The Cancer Imaging Archive (TCIA) [15, 16]. Patient characteristics, including age, gender, pathological grade, tumor-node-metastasis (TNM) stage, and follow-up data, were obtained from TCIA. Histological grade was classified as low (grades 1–2) and high (grades 3–4) [12, 17].
Figure 1 illustrates the recruitment pathway for patients in this study. A total of 177 patients with ccRCC were included, with 142 patients in the training group and 35 patients in the testing group at a randomization ratio of 8:2. Inclusion criteria consisted of the following: (1) patients diagnosed with ccRCC, (2) patients who underwent CT-enhanced scans, and (3) availability of complete genetic and clinical information. Exclusion criteria included the following: (1) patients without nephrographic phase CT images and (2) poor-quality CT images.
Fig. 1
The flow diagram of the study
Segmentation and the extraction of radiomics featuresThe nephrographic phase CT images were employed for radiomics feature extraction [12]. Layer-by-layer delineation of the volume of interest (VOI) was performed using ITK-SNAP software (version 3.8, www.itksnap.org/) by two radiologists, each having over 5 years of experience in diagnostic abdominal imaging. The radiologists were blinded to the patients’ pathological grade. A total of 1834 radiomics features were extracted in Python (version 3.6.0) using PyRadiomics (version 3.0.1) from the VOI for each patient with ccRCC. The reliability of the radiomics features was assessed by calculating inter- and intra-class correlation coefficients (ICCs). Radiomics features with ICCs > 0.75 were deemed reliable. For additional details on the ICC analysis, please refer to the Supplementary Material.
DL feature extractionIn this study, a three-dimensional (3D) DL model using the 3D ResNet50 architecture was employed. The VOI was selected as the original image and resized to 96 × 96 × 96 to align with the network’s input size. The model training process consisted of updating the network weights using a cross-entropy loss function, which was utilized for the prediction task. The 3D DL model was then used to extract DL features from each VOI. For each patient in the training and testing groups, a total of 1024 DL features were extracted from the penultimate fully connected layer. All were run in Python (version 3.6.8). We used the PyTorch framework to train the model on NVIDIA RTX 3070 Ti graphics processing units. The network optimization was performed using the Adam optimizer with a learning rate of 0.001. The training process spanned 300 epochs, with a batch size of 4.
Functional enrichment analysisTranscriptomic data from 142 ccRCC patients were obtained from the TCGA database for genetic analysis. The differential expression of genes (DEGs) between high-grade and low-grade ccRCC samples was analyzed using the “DEseq2” package in R software. Subsequently, a Gene Ontology (GO) enrichment analysis was conducted on the DEGs to identify biological processes, cellular components, and molecular functions that exhibited significant enrichment in one group compared to the other.
Radiomics, DL, and transcriptomics feature selection and models buildingThe analysis proceeded in three main steps. First, univariate regression analysis was employed to identify the radiomics, DL, and transcriptomics features that were significantly associated with grade and prognosis. Second, the least absolute shrinkage and selection operator (LASSO) method was applied to the training group in order to select the most important features. Finally, the selected important features were utilized to construct the radiomics, DL, and transcriptomics models.
Performance of the three models and multi-modelFigure 2 illustrates the workflow encompassing the fundamental steps in radiomics development. A multi-model was created by integrating radiomics, DL, and transcriptomics models through logistic regression. To evaluate the performance of these models, metrics such as the area under the receiver operating characteristic (ROC) curve (AUC), calibration curve, and decision curve analysis (DCA) were utilized for both the training and testing datasets.
Fig. 2
The workflow of the basic steps in multi-model development
Survival analysis and immune cells infiltration analysisPatients were initially stratified into high-risk or low-risk groups using the median scores obtained from the multi-model. Subsequently, follow-up data was analyzed to determine progression-free survival (PFS) and overall survival (OS) outcomes. PFS was defined based on the occurrence of new tumor events, including disease progression, local recurrence, distant metastasis, or death, while OS was calculated from the date of disease diagnosis until either death or the specified cut-off date for follow-up. To visually represent the survival status of the high-risk and low-risk patient groups, Kaplan–Meier plots were generated. The prognostic potential of the multi-model and the survival status of the patients were evaluated using Harrell’s concordance index (C-index).
Enrichment scores for specific immune cells in ccRCC were calculated using Single Sample Gene Set Enrichment Analysis (ssGSEA) in R software for each patient. Additionally, a comparison of the enrichment scores of immune cell infiltration was performed between high-risk and low-risk patients. This analysis aimed to examine the association between the multi-model and histological grade, shedding light on the relationship between the predictive model and immune cell composition.
Statistical analysisContinuous variables that exhibited a normal distribution were reported as mean and standard deviation. Categorical variables were compared using chi-square tests, while independent samples t-test or Mann–Whitney U test was utilized to compare continuous variables. Statistical significance was considered when the p value was less than 0.05. The statistical analyses were conducted using Python (version 3.6.8) and R software (version 4.2.2).
Comments (0)