
記住我






GCT account for 1–2% of all neoplasms occurring in the male population, but represent the most common cancer type in young men [1]. The CXCR4 / CXCR7 / CXCL12-cascade has been postulated to play a major role during metastasis in GCT. In this study, we therapeutically targeted this axis in GCT. Further, we characterized the molecular role of this cascade.
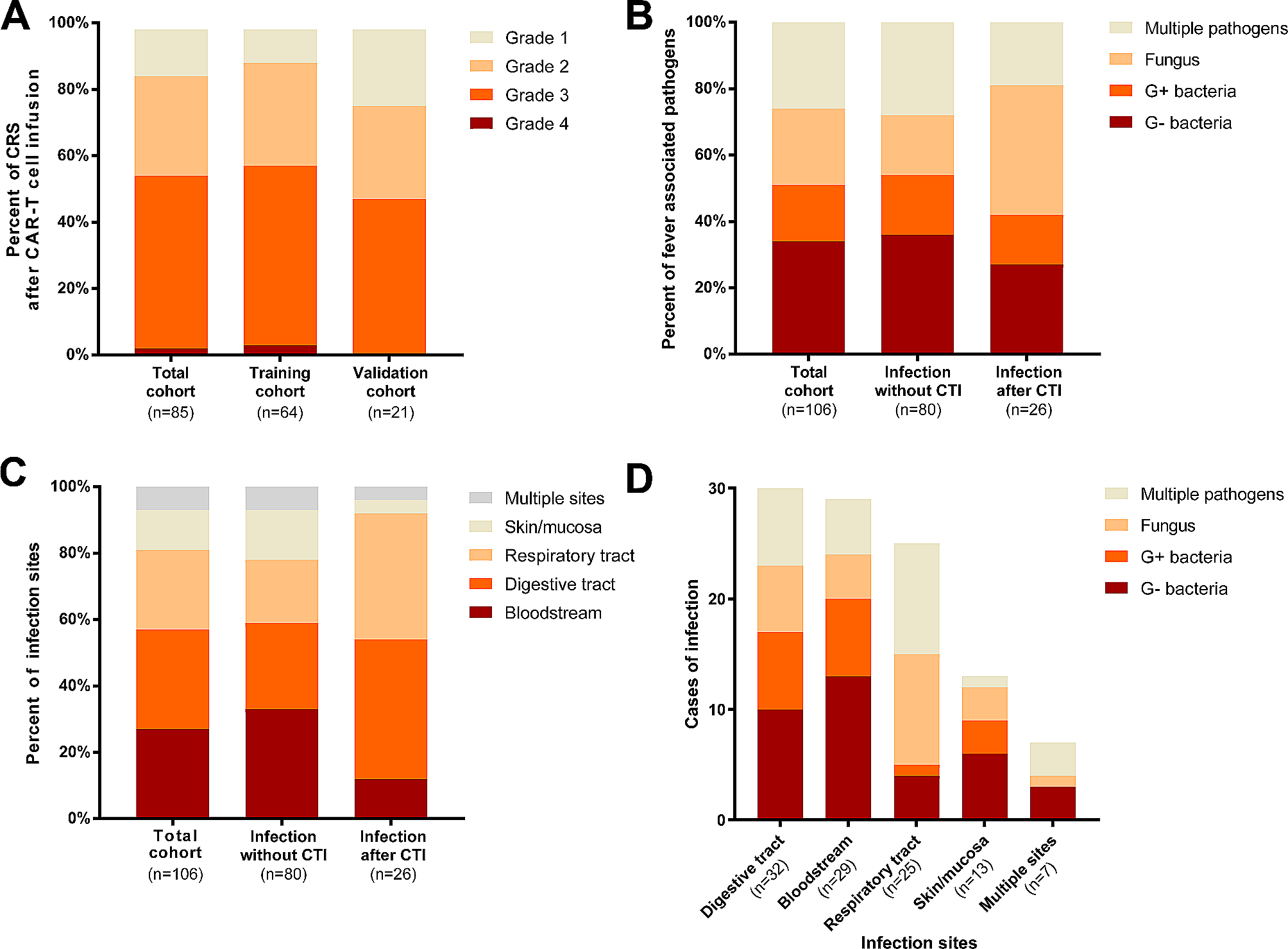
According to ‘The Cancer Genome Atlas’ (TCGA), mutations in CXCR4 / CXCR7 were not found in GCT (Fig. S1A). On protein level, seminoma (SEM) tissues presented as CXCR4+/ CXCR7−, embryonal carcinoma (EC) as CXCR4− / CXCR7−, choriocarcinoma (CC) as CXCR4− / CXCR7+, and yolk-sac tumors (YST) as CXCR4+ / CXCR7+ (Fig. 1A). In mixed GCT (YST + EC), only YST cells were CXCR4+ / CXCR7+. In GCT cell lines including cisplatin-resistant subclones (-R), similar observations were found on mRNA level (SEM: TCam-2CXCR4+/CXCR7+; EC: 2102EP / NT2/D1 / NCCITCXCR4−/CXCR7−; CC: JAR / JEG-3 / BeWoCXCR4−/CXCR7+; YST: GCT72CXCR4+/CXCR7+; intermediate EC / YST: 1411HCXCR4+/CXCR7+) (Fig. 1B; Fig. S1B). CXCR7 expression profiles could be confirmed on protein level (Fig. 1C). Previously, on protein level we already demonstrated that SEM and YST cells present as CXCR4+, while EC and CC cells were CXCR4− [2]. Fibroblasts (MPAF) showed negligible CXCR4 / CXCR7 levels, while expressing and secreting CXCL12 (Fig. 1B; Fig. S1C).
Fig. 1
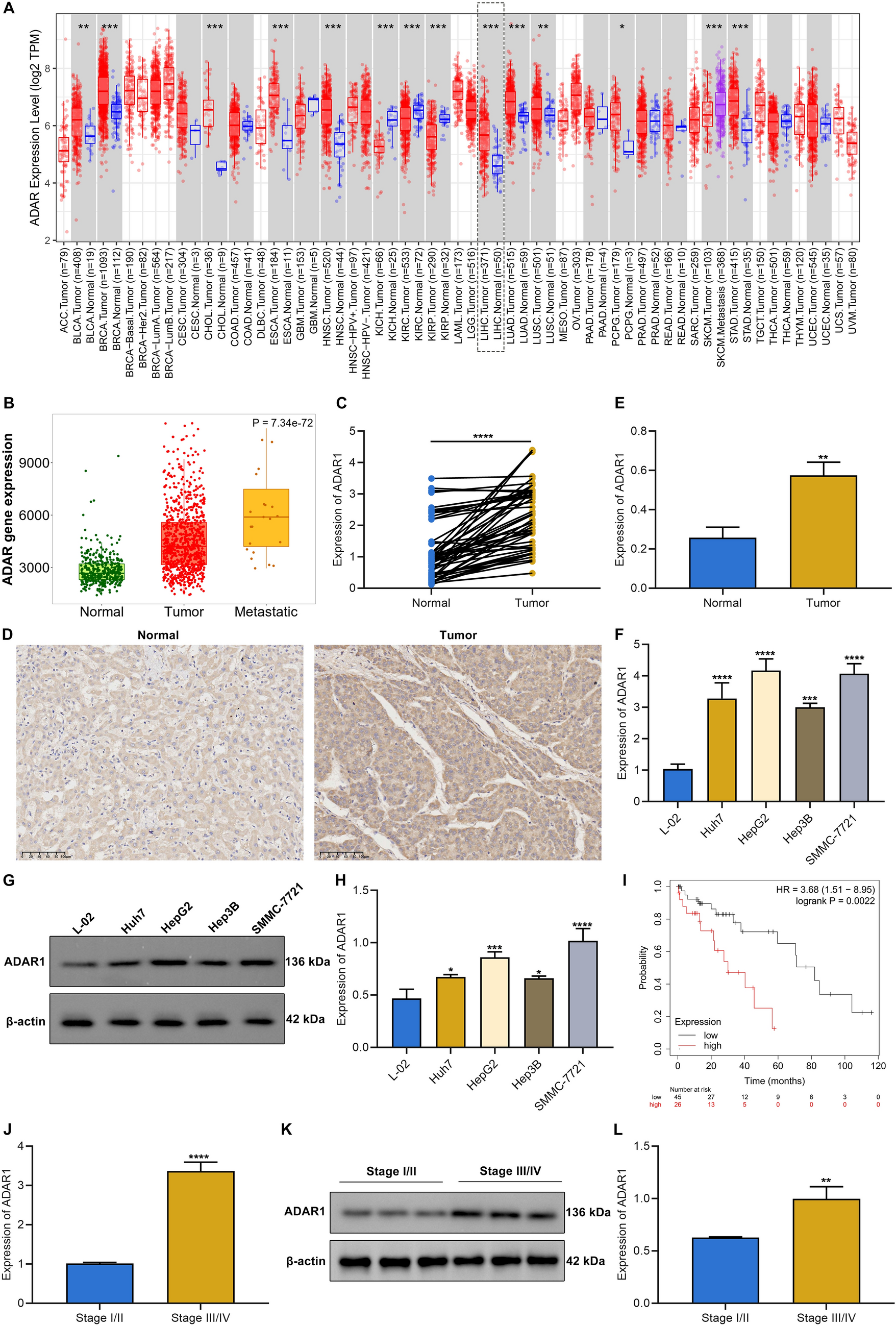
(A) Immunohistochemical evaluation of CXCR4 / CXCR7 in SEM, CC, YST and YST + EC mixed GCT. (B) Relative expression of CXCR4, CXCR7, and CXCL12 in GCT cell lines and fibroblasts (MPAF). ACTB and GAPDH were used as housekeeping genes. (C) Flow cytometry data of CXCR7-APC stained GCT cell lines (blue) compared with unstained controls (grey). (D) XTT cell viability assays of GCT cell lines and fibroblasts (MPAF) treated with CXCR4-NDC or CXCR4-NB alone for 24–96 h. (E) LD50 values measured by XTT cell viability assays 72 h after treatment with cisplatin (CisPt, µM) or CXCR4-NDC (nM) and color-coded changes in cell cycle distribution (mitotic catastrophe = red; changes < 5% = grey) upon treatment with CXCR4-NDC (LD50 concentrations) for 72 h as compared to the CXCR4-NB alone. (F) Lollipop graph summarizing the relative number of apoptotic cells in CXCR4+ GCT cells and CXCR4− MPAF upon treatment with either CXCR4-NDC or CXCR4-NB alone (LD50 concentrations) for 72 h
We therapeutically targeted CXCR4 by a nanobody coupled to the spindle-toxin monomethyl-auristatin-E (nanobody-drug-conjugate; NDC) [3]. Treatment of SEM and YST cells with the CXCR4-NDC decreased cell viability compared to the uncoupled CXCR4-nanobody (CXCR4-NB) (Fig. 1D, E). The LD50 concentrations for the CXCR4-NDC were between 56.4 and 310.6 nM for GCT cells (including cisplatin-resistant sub clones (-R)) and > 500 nM for fibroblasts, thereby opening a therapeutic window (Fig. 1E). A CXCR4-NDC treatment mainly resulted in mitotic catastrophes (TCam-2 / -R, 1411H, BeWo) and induction of apoptosis in CXCR4+-cells (Fig. 1E, F; Fig. S1D). In fibroblasts, the cell cycle and apoptosis remained unaffected (Fig. 1E, F; Fig. S1D).
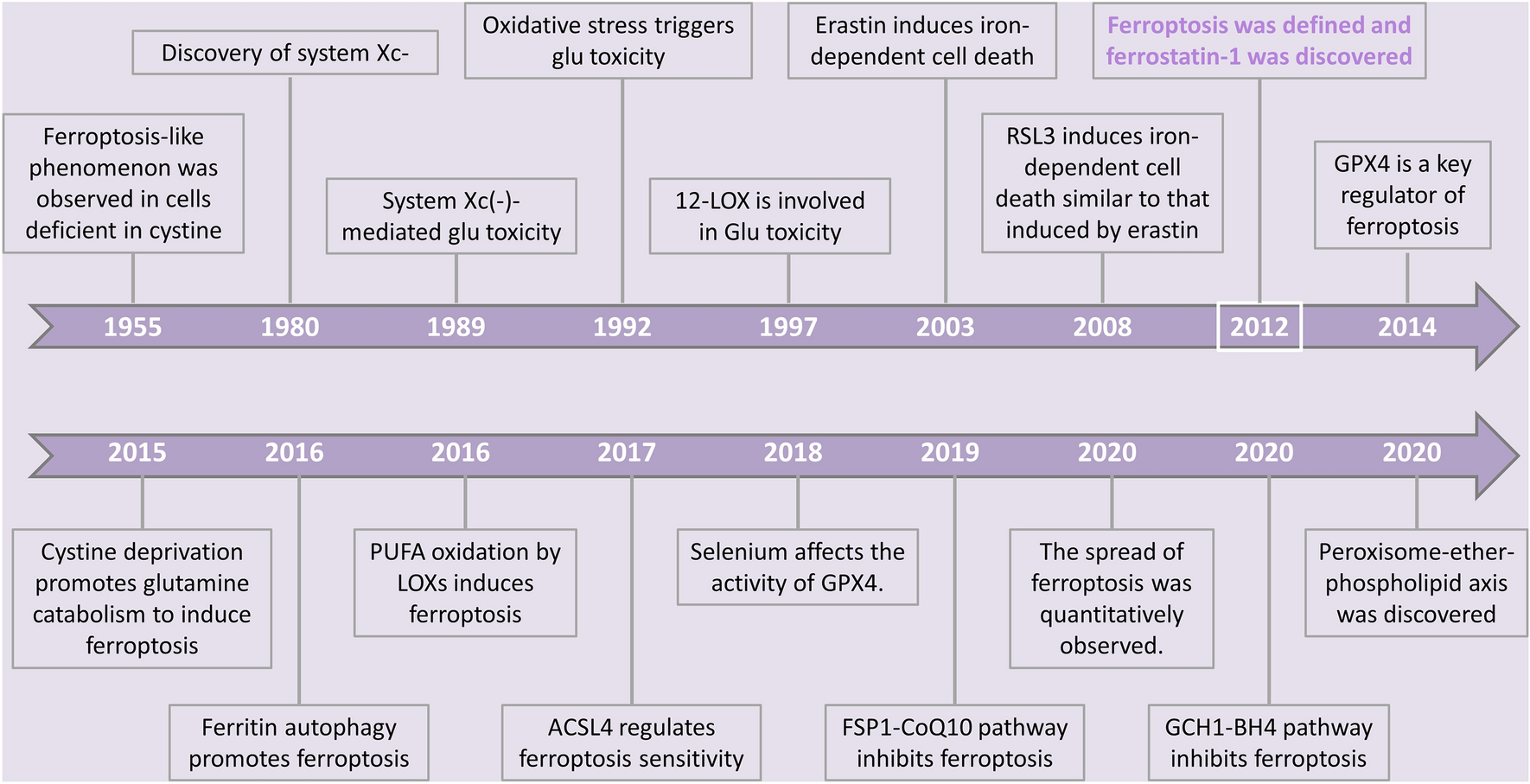
Next, we deciphered the molecular role of this signaling axis on mRNA and phospho-proteome level. Treatment with recombinant CXCL12 (100 ng / ml, 8 h) resulted in enhanced levels of FOS and MMP3 in GCT72CXCR4+/CXCR7+ and BeWoCXCR7+ cells, while BeWo cells further showed increased expression of CD44, IL6, ITGA4A, and MMP1 (Fig. S1E). Further, phospho-kinase-arrays were performed in CXCR4+ / CXCR7+ cells 24 h after treatment with recombinant CXCL12 (250 ng / ml) (Fig. 2A; Fig. S1F). In TCam-2CXCR4+ we found increased phosphorylation (p-) of β-Catenin and STAT5a / b (Y699), in 1411HCXCR4+/CXCR7+ elevated p-GSK-3α / β (S21 / S9), p-p53 (S392) and p-WNK1 (T60), and in BeWoCXCR7+ enhanced levels of p-GSK-3α / β (S21 / S9), p-p53 (S392 / S46), p-SRC (Y419) and p-WNK1 (T60). In GCT72CXCR4+/CXCR7+ cells, phosphorylation of thirteen signaling-molecules, including ERK1 / 2 (T202 / Y204, T185 / Y187), GSK-3α / β (S21 / S9), β-Catenin, and p53 (S15 / S46) was reduced, while p-p53 (S392) was strongly enhanced. Phosphorylation of p53 at S15 has been described to increase stability of p53 by blocking MDM2 binding [4, 5]. Similarly, phosphorylation of S392 is also known to stabilize p53, influence its mitochondrial translocation and a transcription-independent apoptotic function. At a later time-point of DNA damage, S46 phosphorylation initiates the p53-mediated apoptosis through the induction of pro-apoptotic genes, such as p53AIP1 [4, 5]. Hence, in CXCL12-stimulated BeWo cells, p53 phosphorylation at S15 and S46 might indicate an enhanced and stabilized pro-apoptotic p53 signaling cascade. Vice versa, CXCL12-stimulated GCT72 cells revealed a contrary phosphorylation pattern with diminished S15 and S46 phosphorylation of p53. Generally, the basal phosphorylation of all three evaluated p-p53 sites, particularly S46 and S392, is significantly higher in BeWo cells as compared to GCT72 (Fig. S1F), thereby indicating a more profound apoptosis signaling in BeWo cells upon treatment with the CXCR4-NDC (Fig. 1E, F). Thus, reduced p-p53-S15 / S46 after CXCL12-stimulation might diminish the activity of the apoptotic cascade in YST cells, thereby contributing to the high resistance of YST cells to cisplatin.
Fig. 2
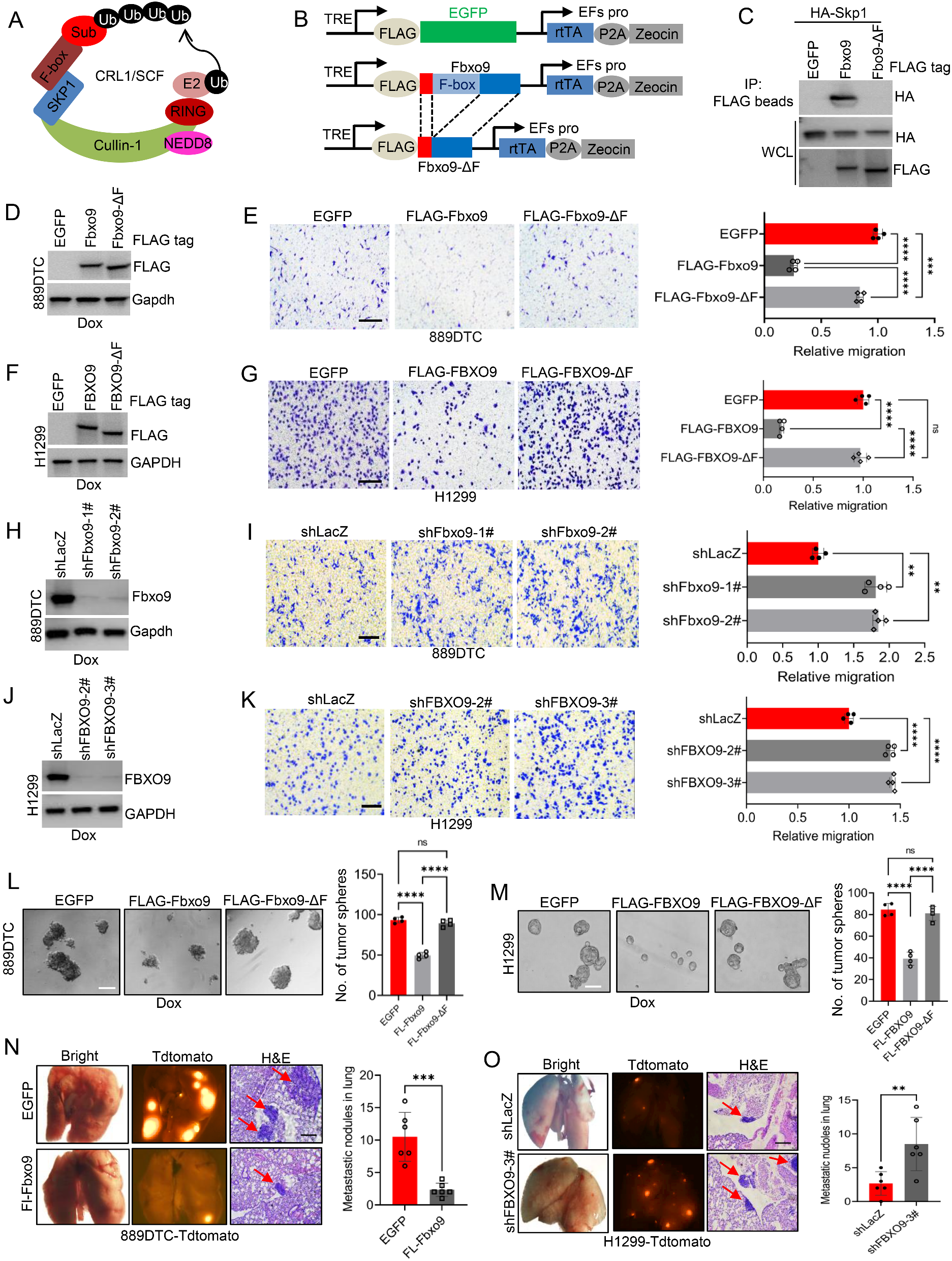
(A) Densitometric evaluation of relative pixel intensities (normalized to untreated controls) of the indicated phosphorylation sites in cell lysates from GCT72, 1411H, TCam-2 and BeWo cells treated with recombinant CXCL12 (250 ng / ml) for 24 h, as measured by a human phospho-kinase array. (B) Relative migration of GCT72 and 1411H cells treated with either 100 ng / ml recombinant CXCL12, 100 nM CXCR7-NB (VUN702), or the combination of both, in comparison with the untreated control. (C) Box plot summarizing the number of proliferative GCT72 cells treated with 100 ng / ml recombinant CXCL12, 20 µM CXCR4-inhibitor AMD3100, or the combination of both for 24 h in comparison to the untreated control. (D) Box plot summarizing the number of proliferative 1411H cells treated with 100 ng / ml recombinant CXCL12, CXCR4-inhibitors WZ811 (5 µM) / LY2510924 (50 nM), or the combination of both for 32 h in comparison to the untreated control. (E) Densitometric evaluation of western blot data of phospho- and total-ERK in EC cells (2102EP, NCCIT, NT2/D1) treated daily with 100 nM ERK inhibitor SCH772984 for 96 h. (F) Relative mRNA expression of CXCR4 and CXCR7 in EC cell lines (2102EP, NCCIT, NT2/D1) treated daily with 100 nM SCH772984 for 96 h as compared to untreated controls. ACTB and GAPDH were used as housekeeping genes. (G) Model summarizing key findings related to the CXCR4 / CXCR7 / CXCL12 axis. SEM present as CXCR4+ / CXCR7−, YST as CXCR4+ / CXCR7+, CC as CXCR4− / CXCR7+ and EC as CXCR4− / CXCR7−. SEM (also with occult YST subpopulations), YST and CC are targetable by CXCR4- and / or CXCR7-NDC, respectively. CXCL12 stimulated CXCR4 / CXCR7 enhanced proliferation and migration in YST cells. In EC cell lines, inhibition of MAPK (ERK1 / 2) signaling allows for re-induction of CXCR7 expression (and partly CXCR4)
Furthermore, the migratory and proliferative capacity of GCT72CXCR4+/CXCR7+ and 1411HCXCR4+/CXCR7+ cells was enhanced upon CXCL12-stimulation; this effect could be blocked upon application of a CXCR7-blocking nanobody (CXCR7-NB) or CXCR4-inhibitors (AMD3100, WZ811, LY2510924) (Fig. 2B-D, G). The CXCR4 / CXCR7-function after CXCL12 stimulation might also be mediated by receptor-heterodimerization [6, 7]. In YST CXCR4+/CXCR7+ cells, we observed that blocking CXCR4 / CXCR7, abolished the effects on proliferation and migration, suggesting that both receptors act in concert to mediate their molecular functions.
We noted a discrepancy between a described CXCL12-dependent MAPK activation and our observed decreased MAPK signaling in CXCL12-stimulated cells [8,9,10]. Thus, we questioned, if ERK1 / 2 inhibition would influence CXCR4 / CXCR7 expression in ECCXCR4−/CXCR7− cells. Indeed, the ERK1 / 2 inhibitor SCH772984 decreased levels of total- and p-ERK1 / 2 (T202 / Y204-T185 / Y187), while increasing CXCR7 expression (and CXCR4 in NT2/D1) (Fig. 2E, F; Fig. S1F). Thus, in EC cells, CXCR7 expression seems to be suppressed by MAPK-signaling, while in YST, diminished MAPK signaling after CXCL12 stimulation seems to allow for CXCR7 expression (Fig. 2G).
In summary, we highlighted the CXCR4-NDC as a therapeutic option for CXCR4+ SEM and YST (Fig. 2G). In YST and CC, also CXCR7 is a putative target, since an uncoupled CXCR7-NB was able to block the CXCR7-mediated molecular effects and proved to be functional. Thus, using a combination of CXCR4- and CXCR7-NDC could be advantageous for the treatment of mixed GCT consisting of SEM, YST and CC. The ability to target YST is of paramount interest, since YST represent the most aggressive GCT entity responsible for a majority of GCT-related death. Under chemotherapy, development of YST represents an escape mechanism, resulting in therapy-resistant relapses. Additionally, about 5% of metastatic SEM present as aggressive relapses accompanied by elevated AFP level, pointing at a YST-subpopulation [11]. In this setting, using a CXCR4-NDC would be beneficial to target both, SEM and the occult YST cells, by rendering the therapy more efficient and reducing the risk of a relapse. Due to tumor heterogeneity, the clinical need to explore tumor subtype-specific targets remains. As such, we identified CD24 to be a specific target for the treatment of EC using NK-CD24-CAR cells [12]. Moreover, we identified the tight-junction molecule CLDN6 as a putative target for the treatment of SEM, EC, CC, and partly YST by using a CLDN6-antibody-drug-conjugate [13]. As such, depending on the tumor subtype, a combined (immuno)therapeutic option should be considered for the treatment of heterogeneous tumors.
留言 (0)