
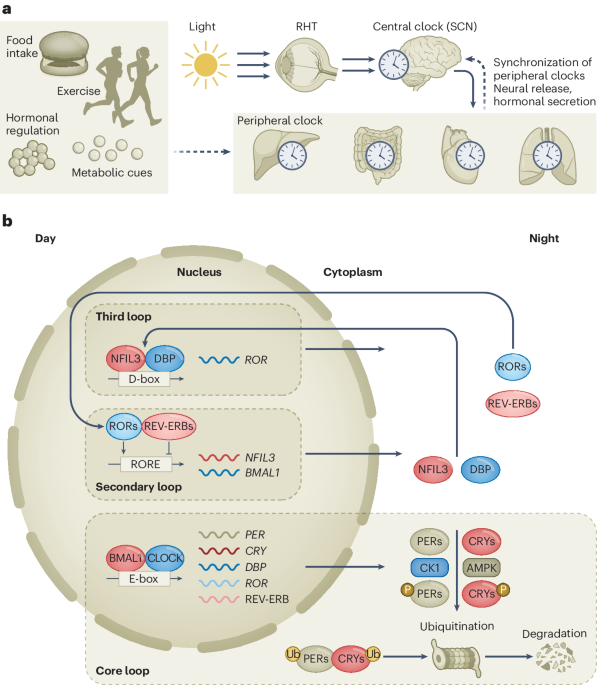
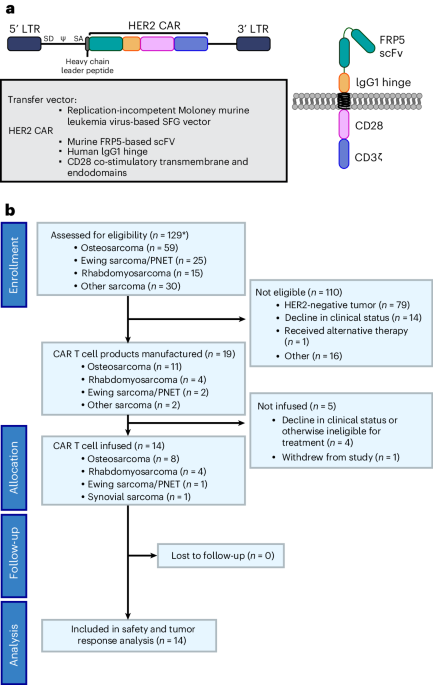
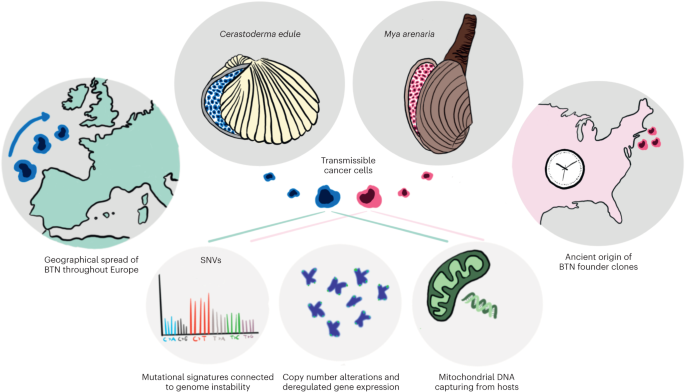
記住我

To generate a representative AML PDX inventory, primary bone marrow or blood samples from 50 patients were tested for engraftment and development of AML in NOD/SCID/IL2gR−/−/hIL3,CSF2,KITLG (NSGS). The overall success rate for primary engraftment in NSGS was 70%, defined by bone marrow, spleen or peripheral blood donor chimerism of at least 20%, splenomegaly (spleen weight >70 mg), anemia (HCT < 35%) or thrombocytopenia (PLT < 400 × 106 ml−1), microscopically visible AML infiltration into the spleen or liver and peripheral blood blast morphology (Extended Data Fig. 1a–n). Successfully engrafted NSGS recipients developed AML with a median onset of 173 d post-transplant (Extended Data Fig. 1p).
From the individual samples from patients with AML that successfully engrafted in NSGS, 30 were randomly selected and characterized based on clinical parameters, including patient age, sex, ELN2017 risk, World Health Organization (WHO) disease classification and molecular profiles obtained by transcriptional and mutational sequencing (Fig. 1a–c). All ELN2017 prognostic risk (favorable, intermediate and adverse) and age categories were represented; 17 samples were from female and 13 samples from male AML patient donors (Fig. 1b). Oncogenic mutations were most frequently detected in NPM1, DNMT3A and FLT3 loci and overall, this AML PDX resource recapitulated the genetic abnormalities that are observed in large clinical AML cohorts2 (Fig. 1c).
Fig. 1: Integrative analysis of samples from patients with AML.
a, Unsupervised hierarchical clustering analysis on the expression of 300 transcripts with the greatest variance-to-mean ratios among 30 individual AMLs from our repository that can successfully generate AML PDX. MLD, multi-lineage dysplasia; MDS, myelodysplastic syndromes; MLL, mixed-lineage leukemia; NOS, not otherwise specified. b, Key clinical characteristics of patients from whom AML samples were derived including age at diagnosis, sex, ELN2017 prognostic risk group and WHO class of disease. c, OncoPrint of the most frequently detected mutations in AMLs by targeted next-generation sequencing of 585 genes associated with hematological malignancies (the MSKCC HemePACT assay)31.
A phase II-like preclinical trial of imetelstat in AML PDXTo test the preclinical efficacy of imetelstat in AML, the characterized 30 individual samples from patients with AML were each transplanted into 12 NSGS recipients (n = 360 PDXs in total). Once AML burden was detected, PDXs were randomized and treated with imetelstat or vehicle control (PBS) until disease onset or a survival benefit of at least 30 d was reached. Median survival was significantly prolonged in imetelstat compared to PBS-treated PDXs (155 d versus 100 d after start of treatment, P < 0.0001; Fig. 2a). AML burden measured as peripheral blood donor chimerism per day was significantly lower in imetelstat compared to vehicle-treated recipients (Fig. 2b). Moreover, end point peripheral blood donor chimerism, bone-marrow cellularity and donor chimerism as well as the absolute number of AML patient-derived cells were significantly reduced in recipients treated with imetelstat when compared to vehicle control (Fig. 2c–f). Furthermore, imetelstat treatment significantly reduced splenic AML donor chimerism (Fig. 2g). We next assessed AML surface marker expression associated with leukemia-initiating activity11,12,13 (Fig. 2h). Imetelstat significantly diminished the CD34+CD38− leukemic stem cell-enriched splenic AML cell population (Fig. 2i). In normal human hematopoiesis using two independent CD34-enriched cord blood xenografts in NSG recipients, the effects of imetelstat were predominantly seen in B lymphocytes with relative preservation of the myeloid and stem cell populations (Extended Data Fig. 2a–q).
Fig. 2: The efficacy of imetelstat in a randomized phase II-like preclinical trial in AML PDX.
a, Two-tailed Kaplan–Meier survival analysis of vehicle control (PBS; n = 180) or imetelstat-treated (n = 180) AML PDX. P < 1 × 10−4 according to Gehan–Breslow–Wilcoxon. b–g, Analysis of AML disease parameters. Peripheral blood (PB) donor chimerism area under the curve (AUC) per day (b), end point PB donor chimerism (c), bone-marrow (BM) cellularity (d), BM chimerism (e), the number of AML donor-derived cells in PDX BM (f) and splenic (SPL) donor chimerism (g). h,i, Flow cytometric analysis of AML surface marker expression CD34, CD38 and GPR56. Gating strategy (h). The percentage of CD34+CD38− viable CD45+ SPL singlets (i). Data are presented as median ± 95% confidence interval (CI) (b–g,i). Statistical analysis was performed on log-transformed data using an unpaired two-sided t-test, considering detection limits at 1 × 10−3. P = 2.21 × 10−10 (b), P = 7.79 × 10−8 (c), P = 1.37 × 10−4 (d), P = 7.32 × 10−3 (e), P = 8.82 × 10−5 (f), P = 1.83 × 10−3 (g), P = 7.44 × 10−5 (i). Asterisks (*) denote statistically significant comparisons with P < 5 × 10−2. j,k, GSEA on RNA-seq data from sorted viable hCD45+ cells collected from imetelstat or PBS-treated AML PDXs. n = 16 AML PDXs per treatment group. Cytoscape nodes represent gene sets with a cutoff of q < 0.1 (j); GSEA on hallmark signatures with the top five enriched signatures highlighted in color (k). l–n, TERT messenger RNA (mRNA) expression results obtained from RNA-seq analysis described as above (l). FC, fold change. Telomere length in viable CD45+ SPL cells from imetelstat versus PBS-treated AML PDXs measured by qPCR (m) and confirmed by telomeric restriction fragment analysis (n). Statistical analysis (l,m) was based on paired two-tailed t-tests comparing AML PDXs treated with imetelstat (n = 16) or PBS (n = 16). P = 9.48 × 10−2 (l), P > 5 × 10−2 (m). Data are presented as mean ± s.e.m. NS, not significant.
We next aimed to compare imetelstat responses to those obtained with standard induction chemotherapy (cytarabine plus anthracycline) in AML PDX from 20 individual samples from patients with AML in an independent cohort using NOD.Rag1−/−Il2Rg−/−/ hIL3,CSF2,KITLG (NRGS) recipients14. Imetelstat matched the similar benefit conveyed by standard chemotherapy (139 d) comparative to 104 d in the vehicle control group and this was accompanied by significant reductions in peripheral blood AML burden (Extended Data Fig. 3a,b); however, the individual samples from patients with AML could be classified into either preferential imetelstat or preferential chemotherapy responders (Extended Data Fig. 3c). Preferential responses to imetelstat when compared to standard induction chemotherapy were associated with baseline mutations in NRAS, JAK2 or GLI1 (Extended Data Fig. 3d).
We next assessed the transcriptional consequences of imetelstat therapy in a cohort of PDX from eight randomly chosen individual samples from patients with AML in vivo (n = 4 sustained (RBWH-37, −47, −48 and −36), n = 3 intermediate (RBWH-46, −56 and −42) and n = 1 poor (RBWH-44) responders to imetelstat). Gene expression signatures annotated as interferon signaling, cell cycle, transcriptional regulation by TP53 and MAPK signaling were significantly enriched in AML donor cells from imetelstat-treated compared to vehicle control-treated PDX (Fig. 2j,k). TERT messenger RNA expression levels were trend-wise reduced in AML donor cells derived from imetelstat-treated compared to vehicle-treated PDX spleens (Fig. 2l). Notably, telomere lengths were similar between imetelstat-treated compared to vehicle-treated groups (Fig. 2m,n).
A CRISPR/Cas9 screen to identify key effectors of imetelstatTo investigate the mechanism of action of imetelstat in AML in an unbiased manner, we applied the Brunello guide RNA (gRNA) library15 as a positive selection screen to identify gene knockouts that confer resistance to imetelstat. We used NB4 cells as these demonstrated highest sensitivity to imetelstat when compared to 13 other human hematopoietic cell lines (Extended Data Fig. 7a). Half-maximum inhibitory concentration (IC50) values strongly depended on cell density, demonstrating the presence of an imetelstat inoculum effect (Extended Data Fig. 4a)16. Cas9-expressing NB4 cells transduced with the Brunello library or untransduced controls were cultured in the presence of imetelstat concentrations that resulted in substantial cell death (IC98) of the untransduced control cultures but allowed the enrichment of imetelstat-resistant cells in Brunello-transduced cultures over a time course of 45 d in culture (Extended Data Fig. 4b). Vehicle or mismatch control-treated NB4 cells grew exponentially throughout the course of treatment (Extended Data Fig. 4c). Specific guide RNAs were selectively enriched in Brunello-transduced imetelstat-resistant compared to vehicle-treated and input control cultures (Extended Data Fig. 4d–g). Combined RIGER and STARS gene-ranking algorithms identified seven significant hits: fatty acid desaturase 2 (FADS2), acyl-CoA synthetase long-chain family member 4 (ACSL4), translocase of inner mitochondrial membrane 17A (TIMM17A), late endosomal/lysosomal adaptor, MAPK and MTOR activator 1–3 (LAMTOR1, LAMTOR2, LAMTOR3) and myosin regulatory light-chain interacting protein (MYLIP; Fig. 3a). Ingenuity pathway analysis indicated close functional relationships between the seven hits in regulating lipid metabolism, iron/metal ion binding, mitochondrial matrix and lysosome biogenesis and localization (Fig. 3b).
Fig. 3: Identification of key mediators of imetelstat efficacy using genome-wide CRISPR/Cas9 editing.
Brunello CRISPR/Cas9 positive enrichment screen in NB4 cells. a, gRNA enrichment analysis using STARS and RIGER gene-ranking algorithms in n = 3 independent imetelstat-treated biological replicates. Red circles indicate significantly enriched targets (STARS false discovery rate (FDR) < 0.15 and RIGER score >2.0). b, Cytoscape visualization of the ingenuity pathway analysis (IPA)-derived interaction network connecting the identified significantly enriched gRNA targets. c–f, Competition assays of imetelstat- (red) versus vehicle control (PBS; black)-treated Cas9-expressing NB4 (c), MV411 (d), KO52 (e) and TF1 (f) cultures transduced with n = 2 independent sgRNAs targeting FADS2 (top), n = 4 independent sgRNAs targeting ACSL4 (middle) and n = 2 controls (empty vector and gRNA targeting CD33). Three technical replicates per condition from two independent experiments were pooled. Asterisks (*) denote statistically significant comparisons based on distinct 95% CI on mCherry chimerism AUC between imetelstat and PBS-treated cultures. 95% CI (lower limit, upper limit): NB4 FADS2 PBS (444.4, 456.7) versus imetelstat (771.9, 795.9); ACSL4 PBS (320.1, 344.4) versus imetelstat (428.3, 459.6); editing controls PBS (186.0, 197.8) versus imetelstat (185.4, 203.4) (c). MV411 FADS2 PBS (1,253, 1,302) versus imetelstat (1,421, 1,525); ACSL4 PBS (847.8, 1,160) versus imetelstat (1,262, 1,564); editing controls PBS (896.2, 1,030) versus imetelstat (943.8, 1,073) (d). KO52 FADS2 PBS (812.3, 907.0) versus imetelstat (949.8, 1,023); ACSL4 PBS (648.9, 743.3) versus imetelstat (1,020, 1,095); editing controls PBS (635.3, 727.6) versus imetelstat (654.9, 760) (e). TF1 FADS2 PBS (1,280, 1,325) versus imetelstat (1,585, 1,721); ACSL4 PBS (1,381, 1,644) versus imetelstat (2,486, 2,788); editing controls PBS (905.9, 982.8) versus imetelstat (888.2, 993.3) (f).
We next aimed to validate the most significant hits identified (FADS2 and ACSL4) using single guide RNA (sgRNA)-mediated editing in the NRAS wild-type expressing NB4 and MV411 and the NRAS-mutant KO52 (p.G13R) and TF1 (p.Q61P) AML cell lines. Editing was confirmed by TIDE analysis17 and reduced protein levels (Extended Data Fig. 4h–j).
We performed competition assays to confirm that loss-of-function editing of FADS2 or ACSL4 confers competitive growth advantage under imetelstat pressure in all AML cell lines analyzed (Fig. 3c–f). The observed effects were target-specific as a competitive outgrowth under imetelstat pressure was not observed when CD33 (predicted to have neutral effects on cell functions18) knockouts or empty vector controls were used (Fig. 3c–f).
These results demonstrate that loss-of-function editing of FADS2 or ACSL4 confers competitive growth advantage under imetelstat pressure, identifying ACSL4 and FADS2 as mediators of imetelstat efficacy in AML.
Imetelstat is a potent inducer of ferroptosisACSL4 and FADS2 encode key enzymes regulating polyunsaturated fatty acid (PUFA)-containing phospholipid synthesis. FADS2 is a key enzyme in a lipid metabolic pathway that converts the essential fatty acids linoleate (18:2n6) and α-linolenate (C18:3n3) into long-chain PUFAs19. Targeted lipidomics analysis on 593 lipid species and their desaturation levels20 demonstrated clear effects of imetelstat treatment and FADS2 editing on the cellular lipidome, with imetelstat-treated empty vector control AML cells showing greatest difference to vehicle-treated empty vector control and FADS2-edited cells. (Extended Data Fig. 5a). Moreover, we found a significant enrichment of phospholipids containing fatty acids with three unsaturated bonds in imetelstat-treated compared to vehicle control-treated NB4 cells and this enrichment of lipid desaturation was diminished by FADS2 editing (Fig. 4a and Extended Data Fig. 5b). Moreover, imetelstat increased the levels of phospholipids with triglycerides and reduced the levels of phospholipids containing cholesteryl esters and ceramides when compared to vehicle control in an FADS2-dependent manner (Extended Data Fig. 5c). Taken together, these data demonstrate imetelstat-induced PUFA phospholipid synthesis in an FADS2-dependent manner.
Fig. 4: Imetelstat is a potent inducer of ferroptosis.
a, Lipid desaturation analysis of FADS2-edited (FADS2-sg1 and FADS2-sg2) or non-edited (empty vector control) NB4 cells treated with imetelstat (4 μM at a seeding density of 2.5 × 105 cells per ml culture) or vehicle control (PBS) for 24 h. The graph depicts the median log2FC of the number of total unsaturated bonds in lipid species in the respective comparisons outlined in the legend. Shading represents the 95% CI. n = 3 replicates from distinct cell passages and independent experiments. b,c, CellROX Green (b) and C11-BODIPY (c) analysis in ACSL4-edited (n = 4 independent gRNAs), FADS2-edited (n = 2 independent gRNAs) or non-edited (n = 2 independent replicates, Cas9, empty vector) NB4 or MV411 cell lines treated with imetelstat (4 μM) or PBS. Time points of analysis were 24 h (NB4) and day 4 (MV411). Three technical replicates per condition were pooled. Data are presented as mean ± s.e.m. One-way analysis of variance (ANOVA) was used and adjusted for multiple comparisons. NB4, P < 1 × 10−4 (non-edited + PBS versus non-edited + imetelstat), P = 2 × 10−4 (non-edited + imetelstat versus ACSL4-edited + imetelstat), P = 2 × 10−4 (non-edited + imetelstat versus FAD2S-edited + imetelstat); MV411, P < 1 × 10−4 (non-edited + PBS versus non-edited + imetelstat), P < 1 × 10−4 (non-edited + imetelstat versus ACSL4-edited + imetelstat), P < 1 × 10−4 (non-edited + imetelstat versus FADS2-edited + imetelstat) (b). NB4, P < 1 × 10−4 (non-edited + PBS versus non-edited + imetelstat), P < 1 × 10−4 (non-edited + imetelstat versus ACSL4-edited + imetelstat), P < 1 × 10−4 (non-edited + imetelstat versus FADS2-edited + imetelstat); MV411, P < 1 × 10−4 (non-edited + PBS versus non-edited + imetelstat), P < 1 × 10−4 (non-edited + imetelstat versus ACSL4-edited + imetelstat), P < 1 × 10−4 (non-edited + imetelstat versus FADS2-edited + imetelstat) (c). A repeat experiment was performed that replicated the results.
ACSL4 has been previously identified as key regulator of ferroptosis21. Ferroptosis is a form of cell death that is driven by an imbalance between the production of reactive oxygen species (ROS) during lipid peroxidation and the antioxidant system and may involve autophagic processes depending on the trigger22. A hallmark of ferroptosis is lipid peroxidation, the oxidation of PUFA-containing phospholipids that occurs via a free radical chain reaction mechanism22. Cancer therapies can enhance ferroptosis sensitivity via lipid remodeling that increases levels of peroxidation-susceptible PUFA-containing phospholipids23.
To test whether imetelstat induces lipid peroxidation, we treated various AML cell lines with C11-BODIPY, a fluorescent fatty acid probe that changes its emission spectrum from red to green upon oxidation. In all four AML cell lines tested, imetelstat treatment resulted in a significant increase in mean fluorescence intensity (MFI) of the oxidized fatty acid probe, demonstrating that imetelstat induces lipid peroxidation in AML cells in vitro (Fig. 5b). We next assessed whether also ROS levels were affected by imetelstat. Using CellROX Green to measure ROS production, we found that its MFI was increased by imetelstat and this increase was diminished when the lipid ROS scavenger ferrostatin-1 was added during the incubation step with CellROX Green, demonstrating that imetelstat increases predominantly lipid ROS levels in AML cell lines in vitro (Fig. 5a). Both lipid peroxidation and lipid ROS production were significantly diminished in ACSL4 or FADS2 loss-of function edited AML cell lines, demonstrating that imetelstat-induced lipid peroxidation and lipid ROS production are dependent on functional FADS2 and ACSL4 in vitro (Fig. 4b,c). Pharmacological inhibition of ferroptosis using the lipid ROS scavengers ferrostatin-1 and liproxstatin-1 diminished imetelstat efficacy in all AML cell lines tested (Extended Data Fig. 6). Moreover, the iron chelator deferoxamine mesylate, the 5-lipoxygenase inhibitor zileuton and menadione diminished imetelstat-induced cell death in a substantial proportion of AML cell lines tested (Extended Data Fig. 6).
Fig. 5: Lipid ROS scavenging diminishes imetelstat efficacy.
a,b, CellROX Green (a) and C11-BODIPY (b) flow cytometry on NB4, MV411, KO52 and TF1 treated with imetelstat (4 μM) or vehicle control (PBS). n = 6 replicates pooled from two experiments. Time points of analysis were 24 h (NB4) and day 4 (MV411), day 8 (KO52) and day 5 (TF1). Data are presented as mean ± s.e.m. a, One-way ANOVA was used and adjusted for multiple comparisons. NB4, P < 1 × 10−4 (NB4 PBS versus imetelstat), P = 9 × 10−4 (imetelstat versus imetelstat + ferrostatin); MV411, P = 1 × 10−4 (PBS versus imetelstat), P = 1 × 10−4 (imetelstat versus imetelstat + ferrostatin); KO52, P = 1.84 × 10−2(PBS versus imetelstat), P = 1.95 × 10−2 (imetelstat versus imetelstat + ferrostatin); TF1, P = 6.2 × 10−3 (PBS versus imetelstat), P < 1 × 10−4 (imetelstat versus imetelstat + ferrostatin). b, An unpaired two-sided t-test was used. NB4, P < 1 × 10−4; MV411, P = 1 × 10−4; KO52, P = 9.4 × 10−3; TF1, P < 1 × 10−4. c, C11-BODIPY and ACSL4 messenger RNA (mRNA) analysis on sorted viable CD45+ splenic cells from imetelstat- compared to PBS-treated PDXs from the preclinical trial presented in Fig. 2. C11-BODIPY data (n = 9 PDXs from three individual AML samples with three PDXs per patient sample) are presented as mean ± s.e.m. ACSL4 mRNA data (n = 6 PDXs from the same three individual AML samples with two PDXs per patient sample) are presented as violin plots. Statistics are based on an unpaired two-sided t-test: P < 1 × 10−4 (MFI C11-BODIPY, top), P = 2.145 × 10−1 (MFI C11-BODIPY, bottom); P = 1 × 10−4 (ACSL4, top), P = 9.53 × 10−1 (ACSL4, bottom). d–f, AML PDX treated with vehicle, liproxstatin-1, imetelstat or a combination of liproxstatin-1 with imetelstat for 2 weeks. n = 12 PDX per treatment group. C11-BODIPY (d) and CellROX (e) flow cytometry on splenic CD45+ singlets. PB chimerism (f) at the end of treatment. Data are presented as mean ± s.e.m. (d–f). One-way ANOVA was used and adjusted for multiple comparisons. P = 2.7 × 10−3 (vehicle versus imetelstat), P = 1 × 10−3 (imetelstat versus imetelstat + liproxstatin-1) (d). P = 6.4 × 10−3 (vehicle versus imetelstat), P = 1.934 × 10−1 (imetelstat versus imetelstat + liproxstatin-1) (e). P = 3.3 × 10−3 (vehicle versus imetelstat), P = 4.21 × 10−2 (imetelstat versus imetelstat + liproxstatin-1) (f).
In AML PDXs in vivo, imetelstat-induced lipid peroxidation was associated with increased ACSL4 expression (Fig. 5c). To investigate whether lipid ROS and lipid peroxidation are essential for imetelstat’s mechanism of action in AML PDXs in vivo, we treated AML PDXs with either vehicle control, imetelstat (15 mg kg−1 three times per week), liproxstatin-1 (15 mg kg−1 twice daily) or the combination of both imetelstat and liproxstatin-1 for 2 weeks. Imetelstat-driven lipid peroxidation and ROS production were prevented by liproxstatin treatment (Fig. 5d,e). In vivo liproxstatin treatment diminished imetelstat efficacy in PDXs as measured by peripheral blood AML burden (Fig. 5f).
Taken together, these data provide evidence that imetelstat is a potent inducer of ferroptosis through ACSL4- and FADS2-mediated alterations in PUFA metabolism, excessive lipid peroxidation and oxidative stress.
Lipophagy precedes imetelstat-induced ferroptosisBy integrating transcriptomics and functional genetics, we aimed to investigate the mechanism by which imetelstat induces ferroptosis. We performed an overlay of the in vivo AML PDX RNA-seq datasets from imetelstat and vehicle-treated mice with the Brunello library CRISPR/Cas9 knockout screen data (cutoff criteria of RNA-seq adjusted P value < 0.05 and RIGER P < 0.05) and identified 11 imetelstat target candidates (Fig. 6a). Two of them, VIM (vimentin) and LMNA (lamin A/C), which are part of a common regulatory module (Fig. 6a), have recently been identified as telomeric G-quadruplex-binding proteins24.
Fig. 6: Integrative analysis of transcriptomics and functional genetics.
a, Integration of RNA-seq and CRISPR screen data using relaxed cutoffs (differential gene expression analysis-derived adjusted P < 0.05 and gRNA enrichment analysis-derived RIGER P < 0.05). Thirteen genes (colored dots) passed these cutoff criteria, of which 11 were annotated in ingenuity pathway analysis (IPA) (right). A common regulatory module for VIM, LMNA and RGS18 is highlighted through connecting lines. DEG, differentially expressed gene. b, Confocal microscopy of VIM protein in NB4 cells treated with vehicle control (PBS) or imetelstat for 24 h. Representative images of n = 6 biological replicates. DAPI, 4,6-diamidino-2-phenylindole. c, VIM-editing in NB4 using n = 4 independent sgRNAs. Competition assays of mCherry+VIM-edited cells grown in the presence of mCherry-unedited control NB4 cells, treated with imetelstat (red) or vehicle (PBS) control (black). Plots show data from one representative experiment. Two independent repeats were performed. d, Imaging flow cytometry of lipophagy using C12-BODIPY and LAMP1 in n = 4 independent VIM-edited (VIM-sg1, VIM-sg2, VIM-sg3 and VIM-sg4) or n = 4 independent editing-control (native, Cas9, empty vector or CD33-sg2) NB4 cell lines. Recovery examples of cells showing strong colocalization of C12-BODIPY and LAMP1 indicative of lipophagy activity (top) or cells with weak colocalization indicating insignificant lipophagic flux. Quantification of the percentages of cells with strong colocalization defined as bright detail similarity score >1. Data are presented as mean ± s.e.m. Statistics are based on a one-way ANOVA adjusted for multiple comparisons to PBS-treated editing controls. Editing controls + PBS versus editing controls + imetelstat, P = 1.9 × 10−3; editing controls + PBS versus VIM-edited + PBS, P = 4.955 × 10−1; editing controls + PBS versus VIM-edited + imetelstat, P = 2.128 × 10−1. Comparisons were considered NS when P > 5 × 10−2. Data are from one experiment representative of four independent experiments. This experiment was repeated three times with similar results. e, Chloroquine and imetelstat combination treatments in AML cell lines. Data are presented as mean ± s.e.m. One-way ANOVA was used and adjusted for multiple comparisons. NB4 (n = 3 replicates), P < 1 × 10−4; MV411 (n = 3 replicates), P < 1 × 10−4; KO52 (n = 3 replicates), P = 8 × 10−3; TF1 (n = 2 replicates), P = 5.1 × 10−3; MOLM13 (n = 3 replicates), P < 1 × 10−4; HEL (n = 3 replicates), P < 1 × 10−4. Each experiment was repeated once with similar results.
Recent independent work demonstrated the capacity of imetelstat to form G-quadruplex structures in vitro and this capacity is attributed to the presence of a triple G-repeat (GGG) in its sequence25. These insights prompted us to obtain an additional mismatch control harboring a similar triple G-repeat, but containing enough mismatches to prevent efficient binding to telomerase (Extended Data Fig. 7a). Using an antibody raised against (T4G4)2 intermolecular G-quadruplex DNA structures26,27,28, we found that imetelstat or GGG-containing mismatch but not mismatch 1 significantly interfered with endogenous DNA G-quadruplex structures (Extended Data Fig. 7b). In a panel of 14 human hematopoietic cell lines, GGG-containing mismatch control and imetelstat demonstrated similar efficacies in the majority of AML cell lines tested (Extended Data Fig. 7a). Moreover, GGG-containing mismatch was similarly effective as imetelstat in increasing ROS levels when compared to vehicle control (Extended Data Fig. 7c). Ferrostatin- or deferoxamine mesylate-mediated inhibition of ferroptosis rescued both imetelstat as well as GGG-mismatch-induced cell death (Extended Data Fig. 7d). We next compared the preclinical efficacy of imetelstat with GGG-mismatch and mismatch 1 in an NRAS/KRAS-mutant AML PDX model (RCH-11). In this model, GGG-mismatch was also effective in reducing AML burden (Extended Data Fig. 7e).
In addition to binding telomeric G-quadruplexes, vimentin has long been established as structural component of lipid droplets regulating their biogenesis and stability.
Lipid droplets can undergo selective autophagy (lipophagy) that can result in the induction of ferroptosis29. We hypothesized that imetelstat-induced PUFA phospholipid synthesis, oxidation and ferroptosis can result from lipophagy. Vimentin was highly expressed at protein level in AML cells in vitro (Fig. 6b) and loss-of-function editing of vimentin resulted in a modest competitive growth advantage of AML cells under imetelstat pressure (Fig. 6c). We next assessed lipophagy using C12-BODIPY, a fluorescent fatty acid probe for lipid droplets, in conjunction with the late endosomal marker LAMP1 (ref. 30). Imaging flow cytometry revealed significantly increased colocalization of lipid droplets with the late endosomal marker LAMP1, indicating increased lipophagy (Fig. 6d). To test whether pharmacological inhibition of lipophagy can prevent imetelstat-induced ferroptosis, we cultured AML cells in the presence of imetelstat combined with chloroquine, which inhibits lysosomal hydrolases by increasing the pH and thus lipophagy. Notably, in all AML cell lines tested, chloroquine diminished imetelstat-induced cell death (Fig. 6e).
These results provide evidence for a role of lipophagy-induced ferroptosis in imetelstat’s mechanism of action in AML via impaired lipid droplet homeostasis due to G-quadruplex mediated interference with the structural components of lipid droplets.
Oxidative stress signatures distinguish sustained respondersWe next aimed to identify biomarkers of imetelstat response and resistance. Improved survival in imetelstat-treated AML PDXs correlated with significantly reduced engraftment and disease burden; however, there were clear differences in the magnitude and duration of individual responses (Extended Data Fig. 8). To understand determinants of imetelstat response, we allocated each individual AML patient sample into either sustained, intermediate or poor imetelstat response categories based on the individual effect of imetelstat on AML burden measured in peripheral blood over time (Extended Data Figs. 9 and 10a). All ELN2017 prognostic risk categories were represented in each imetelstat response group, suggesting that the effects observed were not solely explained by favorable disease (Extended Data Fig. 10b). In addition, cytogenetics, sex, age, FLT3-ITD allelic ratio and TERT messenger RNA expression levels at baseline seemed similar among imetelstat response groups (Extended Data Fig. 10c–h).
We next aimed to identify genetic biomarkers of response and resistance to imetelstat therapy by analyzing the data from individual samples from patients with AML at baseline that were generated by genomic sequencing using a comprehensive panel of 585 genes frequently mutated in hematological malignancies31 (Extended Data Fig. 9c). Oncogenic mutations in genes annotated in signaling or cell adhesion/metabolism were trend-wise more frequently observed in sustained compared to poor responders to imetelstat (Fig. 7a and Extended Data Fig. 9c).
Fig. 7: Mutant NRAS and oxidative stress gene expression signatures associate with sustained responses to imetelstat.
Segregation of samples from patients with AML into sustained, intermediate and poor imetelstat response groups based on PB AML burden with n = 14 (sustained), n = 8 (intermediate) and n = 8 (poor). a, Cytoscape visualization of the frequencies of genes with oncogenic mutations (based on the COSMIC database79) in sustained (turquoise), intermediate (light blue) and poor (dark blue) responders to imetelstat. Connecting lines represent co-occurring mutations within the same AML patient sample. b, AML burden in imetelstat-treated normalized to vehicle control-treated PDXs in relation to NRAS mutational status. NRAS wild-type (wt; n = 144 PDXs) and mutant NRAS (mut; n = 36 PDXs). Statistics were conducted according to a two-sided t-test on log-transformed data: P = 2.86 × 10−2. c, Two-tailed survival analysis of PBS and imetelstat-treated AML PDXs divided into groups based on their NRAS mutation status. Median survival was 94 (PBS-treated NRAS-mut; n = 36 PDXs), 389 (imetelstat-treated NRAS-mut; n = 36 PDXs), 100 (PBS-treated NRAS-wt; n = 144) and 153 (imetelstat-treated NRAS-wt; n = 144) days from start of treatment. P = 2.56 × 10−2 comparing imetelstat-treated NRAS-mut to imetelstat-treated NRAS-wt PDX according to Gehan–Breslow–Wilcoxon. d, Cytoscape visualization of GSEA results on RNA-seq data from individual AML patient samples at baseline comparing sustained with poor responders to imetelstat (n = 14 sustained responders; n = 8 poor responders; node cutoff, q < 0.1). Red circles represent gene sets positively enriched in sustained versus poor responders to imetelstat. Blue circles represent negatively enriched gene sets in sustained versus poor responders to imetelstat. e, Hallmark GSEA on RNA-seq data comparing sustained versus poor responders to imetelstat at baseline. The red dotted line represents the cutoff considered for significant enrichment at FDR = 0.25. f, Simple linear regression analysis of baseline telomere length versus imetelstat response in PDXs. n = 30 AML patient samples. P = 7.66 × 10−1; F = 8.998 × 10−1; degrees of freedom numerator, degrees of freedom denominator = 1, 34; slope 95% CI (−2.219 × 10−1, 1.648 × 10−1); y intercept 95% CI (6.023, 8.449); x intercept 95% CI (367.9, +infinity). R2 = 2.639 × 10−3.
Mutant NRAS was associated with enhanced responses to imetelstat therapy. This was evidenced by reduced AML burden and improvement in survival when compared to wild-type NRAS containing AML PDXs (Fig.
留言 (0)