
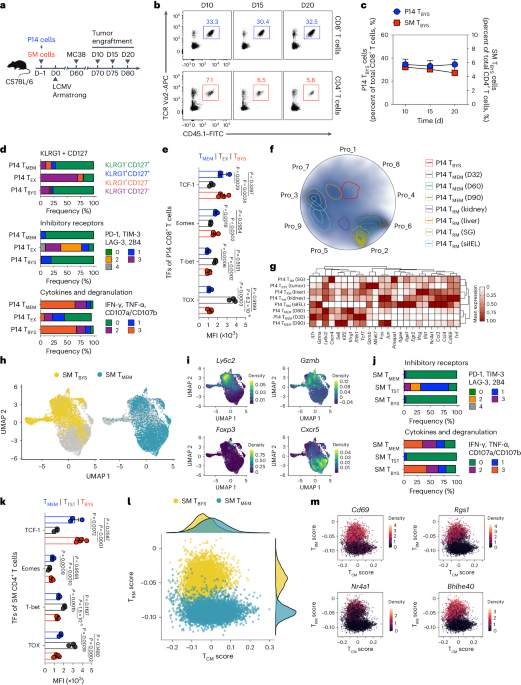
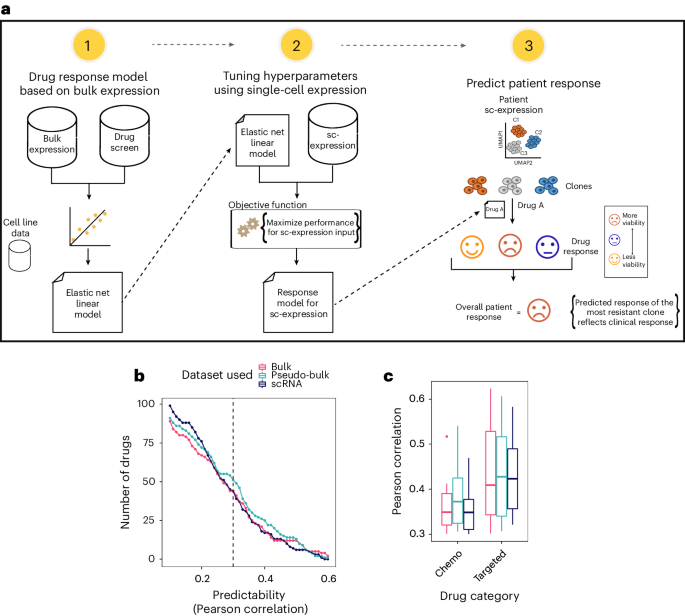
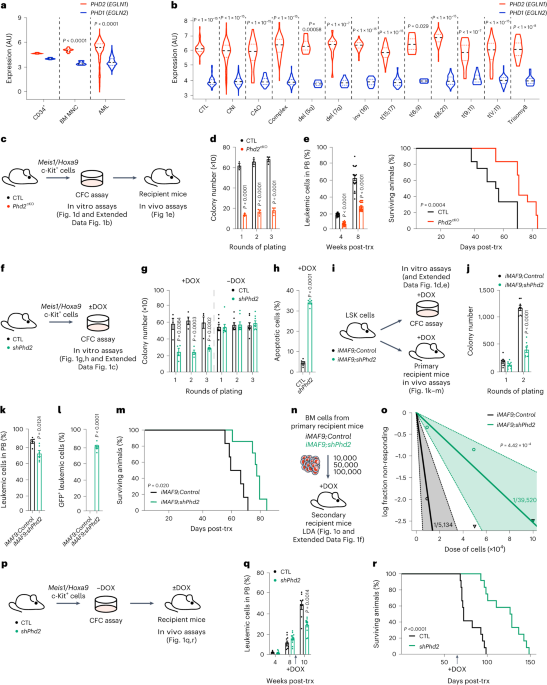
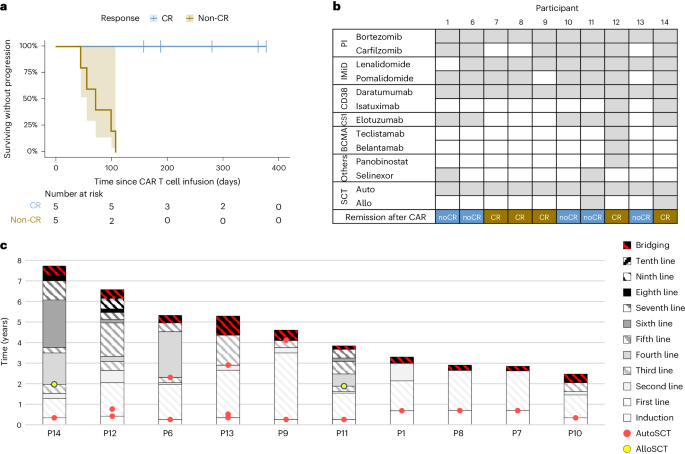
記住我

All primary AML samples used were obtained under Institutional Review Board-approved protocols by the University of California San Francisco (UCSF), Committee on Human Research and following the Declaration of Helsinki. Informed consent was obtained from participants for research purposes, although for this study, all samples were fully deidentified and could not be linked back to relevant clinical information, including participant sex. All mouse experiments were conducted in accordance with an approved protocol by the UCSF Institutional Animal Care and Usage Committee. Mice were housed in the UCSF Animal Care Facility Laboratory Animal Resource Center at the Helen Diller Family Cancer Center at UCSF Mission Bay. Animals were housed in an individual specific pathogen-free suite with up to five mice per ventilated cage and ad libitum access to food and water. The suite was maintained on a 12-h light/12-h dark cycle with controlled temperature (~19–23 °C) and humidity (30–70%) conditions.
Cell lines, PDX and human samplesNomo1 and BV-173 cell lines were obtained from DSMZ, and THP-1, HL-60, MV-4-11, Jurkat, U937, Namalwa and S49.1 cell lines were obtained from ATCC. All cell lines were grown in RPMI-1640 medium (Gibco, 11875093) supplemented with 20% fetal bovine serum (FBS; BenchMark, Gemini, 100-106) and 100 U ml–1 penicillin–streptomycin (UCSF Cell Culture Facility). All cells were grown at 37 °C with 5% CO2. All AML PDXs were procured from the PRoXe at Dana–Farber Cancer Institute under an appropriate materials transfer agreement. Primary AML samples were obtained from the UCSF Hematologic Malignancies Tissue Bank and the Pediatric Hematopoietic Tissue Cell Bank.
Cross-linking and cell surface labelingDSSO-based (Sigma-Aldrich, 909602) XL–MS involving HpHt and PhoX-based (Thermo Fisher Scientific, A52286) XL–MS were each performed with 2.4 × 109 cells (in batches of 6 × 108). Cells were collected and washed (300g, 5 min) three times with PBS. Cross-linker DSSO or PhoX predissolved in DMSO (Sigma-Aldrich, 276855) was then added to the cells at a final concentration of 10 mM and incubated at room temperature for 45 min. Cross-linking was followed by biotinylation of cell surface proteins. Briefly, N-linked sugar residues of the cells were oxidized with 1.6 mM sodium metaperiodate (VWR, 13798-22) for 20 min at 4 °C, followed by installation of biotin on those residues using 10 mM aniline (Sigma-Aldrich, 242284) and 1 mM biocytin hydrazide (Biotium, 90060) for 90 min at 4 °C. The cells were then washed with PBS, snap-frozen and stored at −80 °C until further processing.
Cell surface proteomics sample preparationFrozen cell pellets were thawed on ice and resuspended in 1 ml of RIPA lysis buffer (MilliporeSigma, 20-188) with Halt protease inhibitor (Thermo Scientific, 78430) and 1 mM EDTA (Invitrogen, 15575-038), followed by sonication for lysis. Lysates were centrifuged at 17,000g for 10 min at 4 °C, and clarified supernatant was mixed with 0.5 ml of Neutravidin beads (Thermo Scientific, PI29204), followed by incubation at 4 °C for 2 h in an end-to-end rotor. Beads were washed extensively by vacuum manifold (Promega) with 50 ml of RIPA lysis buffer + 1 mM EDTA, 50 ml of PBS + 1 M NaCl and 50 ml of 2 M urea (VWR, 97063-798) + 50 mM ammonium bicarbonate. Beads were resuspended in 50 mM Tris (pH 8.5) + 4 M urea + 10 mM TCEP (Gold Biotechnology, TCEP10) and 20 mM iodoacetamide (VWR, 97064-926). Ten micrograms of trypsin-LysC (Thermo Scientific, PRV5073) mix was added for on-bead digestion with simultaneous reduction and alkylation. After 2 h, the mixture was diluted to 1.5 M urea using 50 mM Tris (pH 8.5) and incubated overnight (16–20 h). Beads were eliminated by centrifugation, and the resulting supernatant was acidified with 0.5% trifluoroacetic acid (TFA). Peptides were desalted using a SOLA HRP Column (Thermo Scientific, 60109-001) and eluted with 50% acetonitrile (ACN) + 0.1% formic acid (FA).
IMAC purification for PhoXDry peptides were reconstituted in 80% ACN + 0.1% TFA. Superflow Ni-NTA beads were stripped off using EDTA and reloaded with FeCl3 (Sigma-Aldrich, 451649) on a polyprep chromatography column (Bio-Rad, 7326008). Fe3+-loaded beads were transferred to C18 tips (Nest Group, SEM SS18V.25) and incubated for 4–6 min, followed by washing with 0.5% FA. The bound peptides were eluted from beads with 0.5 M potassium phosphate buffer (pH 7.4). Peptides eluted from beads were bound to C18 chromatographic material of the Nest tips, washed three times with 0.5% FA and eluted with 50% ACN + 0.1% FA.
SECSize-based fractionation of peptides was performed using a Superdex Peptide 3.2/300 (GE Healthcare) column and high-performance liquid chromatography (HPLC; Agilent 1260 Infinity II). Dried peptides were reconstituted in the mobile phase of 30% ACN + 0.1% TFA and loaded on the column. Run time was 90 min at a flow rate of 50 µl min–1, and 45 fractions (2 min per fraction) were collected. Fractions associated with the desired molecular weight were dried down in a SpeedVac and stored at −80 °C for MS analysis.
LC–MS and data-dependent acquisition analysisPeptide samples prepared for building the Nomo1 cell surfaceome custom database were loaded on to the EASY-Spray nanocolumn (Thermo Scientific, ES900) installed on a Dionex Ultimate 3000 NanoRSLC instrument coupled with a Q-Exactive Plus mass spectrometer (Thermo Scientific). Peptides were separated primarily over a 313-min gradient ranging from 2.4% to 32% ACN with a flow rate of 0.3 μl min–1. MS scans were performed from m/z 299 to 1,799 at a resolution of 70,000 full-width at half-maximum (FWHM) at m/z 200 with an automatic gain control (AGC) target of 3 × 106 and maximum injection time of 100 ms. The resolution for MS/MS scans was set to 17,500 FWHM at an m/z of 200 with an AGC target of 2 × 105 and maximum collision-induced dissociation of 200 ms. Normalized collision energies of 27%, 30% and 33% in stepped higher collision-induced dissociation mode were used for fragmentation of the top 15 most intense precursor ions with an isolation window of 1.7 m/z. Peptides with a charge state of 2+ or higher were considered for MS/MS. Dynamic exclusion was set to 20 s.
MS-generated .raw files were processed using MSFragger50 within FragPipe v14.0 with default settings unless stated otherwise. Briefly, the spectral data were searched against the human proteome database (UniProt, downloaded 11 May 2021; 20,395 entries). The contaminant and decoy protein sequences were added to the search database using the inbuilt feature of the FragPipe v14.0 pipeline downstream statistical analysis. The inbuilt tools PeptideProphet and ProteinProphet were used for statistical validation for 1% false discovery rate (FDR).
HpHt-based fractionation of DSSO cross-linked peptidesThe SEC fractions 13 and 14, which are enriched with DSSO cross-linked peptides (Extended Data Fig. 1b), were further fractionated by HpHt as described previously23. Briefly, the HpH tip was constructed in a 200-µl pipette tip by packing C8 membrane (Empore 3M) and 5 mg of C18 solid phase (3 μm; Durashell, Phenomenex). The column was sequentially washed with three different solvents/solutions: methanol, ACN and ammonia water (pH 10; 90 µl each). Samples were loaded on the column, followed by washing with 90 µl of ammonia water (pH 10) and a series of ammonia water containing increasing concentration of ACN (6, 9, 12, 15, 18, 21, 25, 30, 35 and 50%). The fractions with 25, 30, 35 and 50% ACN were combined with fractions containing 6, 9, 12 and 21% ACN, respectively, for MS analysis.
LC–MS3 analysis of DSSO cross-linked peptidesThe SEC–HpHt fractions were subjected to LC–MS3 analysis using an UltiMate 3000 RSLC nano-HPLC system coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific), as described previously23. Peptides were separated by reversed-phase LC (50 cm × 75 μm Acclaim PepMap C18 column, Thermo Scientific) with over an 87-min gradient of ACN (4% to 25%) at a flow rate of 300 nl min–1. Initial survey (MS1) scans were measured in the Orbitrap with a scan range from 375 to 1,800 m/z, a resolution of 60,000 FWHM and an AGC target of 4 × 105 with a maximum injection time of 75 ms at top speed per 4 s of cycle time. Ions with a charge of 4+ or greater were selected for MS2 and subjected to fragmentation using collision-induced dissociation with a normalized collision energy of 23%. For MS2 scans, the scan range was set to auto mode, with a resolution of 30,000 FWHM, an AGC target of 5 × 104, a precursor isolation width of 1.6 m/z and a maximum injection time of 100 ms. A targeted inclusion on ions with a mass difference corresponding to the difference in alkene and thiol DSSO fragments (31.9721 Da) was used to select precursors for MS3 analysis. For MS3 scans, higher collision-induced dissociation was used with a normalized collision energy of 28%, the AGC target was set to 2 × 104, and the maximum injection time was set to 125 ms.
Identification of DSSO cross-linked peptidesPeak lists were extracted from the LC–MSn raw files using the in-house software PAVA (UCSF), and the extracted MS3 spectra were searched against a SwissProt database (2021.10.02 version; 20,387 entries) concatenated with its randomized decoy sequences using Protein Prospector (v.6.3.5). The mass tolerances allowed were ±20 ppm for precursor ions and 0.6 Da for fragment ions. The database search was performed with trypsin as a protease with a maximum of three allowed missed cleavages. Cysteine carbamidomethylation was set as the fixed modification. Variable modifications included N-terminal protein acetylation, methionine oxidation and N-terminal conversion of glutamine to pyroglutamic acid. Additionally, three specific modifications resulting from DSSO were included in the search: thiol (C3H2SO, +86 Da), alkene (C3H2O, +54 Da) and sulfenic acid (C3H4O2S, +104 Da)19. The in-house software XL-Tools was used to automatically identify, summarize and validate cross-linked peptides based on Protein Prospector database search results and MSn data. No decoy hits were found after the integration of MS1, MS2 and MS3 data.
Development of the MS3-based XL–MS analysis toolWe developed Ving to assess the MS2/MS3-based cleavable cross-linking database search results to produce a set of cross-linked spectrum matches (CSMs; Extended Data Fig. 1a). Ving is open-source, publicly available software and conceptually based on our previously published search methodology19 but now adapted to the proteomics analysis suite, the TPP. Ving input consists of spectral data in mzML format51 and database search results in PepXML format52.
Ving functions by parsing data to create spectral groups (SGs) consisting of MS2 and MS3 data originating from a single precursor ion. Next, database search results from the MS2 and MS3 scan events, performed using the TPP22 as described previously, are added to each SG. A series of thresholds categorize each SG to determine probable CSMs after peptide sequence assignments to all MS2 and MS3 spectra within all groups. First, MS2 peptide sequence assignments with a probability of >0.8 are labeled as single, non-linked peptide spectrum matches (PSMs). Evidence of an internal lysine residue with a hydrolyzed cross-linker mass refines the classification to dead end or monolinked PSMs. For the remaining SGs, the MS3 peptide assignments and probabilities are evaluated. If the SG has multiple MS3-level peptide sequence identifications with a probability of >0.8 and a cross-linker-modified lysine residue, those sequences are further evaluated as candidate CSMs. If two peptide sequence masses plus the cross-linker mass sum together to match the mass of the original precursor ion, then the group is classified as a CSM. If none of the peptide sequences sum to the precursor mass, then the SG is classified as an incomplete CSM. If the SG has one or zero MS3-level peptide sequences with a probability of >0.8, the group is classified simply as unknown PSMs. After all SGs are evaluated, a human-readable text summary is reported to the user.
LC–MS analysis of PhoX cross-linked peptidesPhoX cross-linked peptide samples were analyzed on a timsTOF Pro mass spectrometer (Bruker Daltronics) as described previously53. Briefly, peptides from each SEC fraction 9–24 (Extended Data Fig. 1c) were loaded on to the column operated using an UltiMate 3000 RSLC nano-HPLC system (Thermo Scientific), and eluted peptides were analyzed with the timsTOF Pro mass spectrometer using a CaptiveSpray source (Bruker Daltonics). Peptides were first trapped on a C18 precolumn (Acclaim PepMap 100, 300 μm × 5 mm, 5 μm, 100 Å; Thermo Scientific), and eluted peptides were subsequently separated on a μPAC 50 column (PharmaFluidics) over 180 min with an ACN gradient ramping up from 3% to 35%, during which the flow rate changed from 900 to 600 nl min–1 for the first 15 min, followed by a constant flow rate of 600 nl min–1.
For MS analysis with the timsTOF Pro mass spectrometer, the mobility-dependent collision energy ramping settings were 95 eV at an inversed reduced mobility (1/k0) of 1.6 V s–1 cm–2 and 23 eV at 0.73 V s–1 cm–2. Collision energies were interpolated linearly between the two 1/k0 values and were kept constant above or below. TIMS scans were not merged, and the target intensity per individual parallel accumulation serial fragmentation precursor ion was kept at 20,000 (with an intensity threshold of 1,000). The mobility range of each scan was kept between 0.6 and 1.6 V s–1 cm–2 with a ramp and accumulation time of 166 ms, and the mass range for MS and MS/MS was set to 100–1,700 m/z. The number of parallel accumulation serial fragmentation MS/MS scans triggered was 14 per cycle (2.57 s), with a maximum of seven allowed precursors per mobilogram. The precursor ion charge states selected for fragmentation ranged between 3+ and 8+, and the isolation width was 2 Th for precursor m/z up to 700 and 3 Th for precursor m/z > 800, in between which it was ramped linearly. The active exclusion was set to 0.4 min (mass width of 0.015 Th and 1/k0 width of 0.015 V s–1 cm–2).
TimsTOF MS data analysisTimsTOF MS data were converted to .mgf format using MSConvert54. The .mgf files were then processed for identification of cross-linked peptides using pLink-2 (ref. 55) with default settings unless stated otherwise. All files were searched against the Nomo1 cell surfaceome-specific custom database (5,280 entries) generated from regular data-dependent acquisition analysis based on prior CSC data (that is, non-cross-linked). For pLink-based cross-linked peptide analysis, trypsin was set as the protease, allowing three missed cleavages. Cysteine carbamidomethylation was set as the fixed modification with methionine oxidation and N-terminal acetylation as the variable modifications. The search was performed with a ±20 ppm mass tolerance window for precursor and fragment ions, and results were reported at 1% FDR.
Flow cytometryImmunostaining of cells was performed as per the instructions from the antibody vendor unless stated otherwise. Briefly, 1 × 106 cells were resuspended in 100 µl of FACS buffer (PBS + 2% FBS) with 1 µg of antibody added. Cells were incubated at 4 °C for 10–15 min and washed three times with FACS buffer. For staining the active form of integrin β2, the antibody incubation step was performed at 37 °C for 1 h. For staining primary AML cells for activated integrin β2, the FACS buffer was RPMI-1640 + 5% FBS + 2% bovine serum albumin (BSA) + 50 µg ml–1 DNase I (Gold Biotechnology, D-301-500). For other primary antibody cell stainings, FACS buffer was D-PBS + 5% FBS + 2% BSA + 5 mM EDTA + 50 µg ml–1 DNase I with Human Trustain (Biolegend, 422302). Compensation used UltraComp eBeads Compensation Beads (Invitrogen, 01-2222-42). All antibodies were diluted 1:20 unless stated otherwise. Flow cytometry analysis was performed on the CytoFLEX platform (Beckman Coulter), and data were analyzed using FlowJo_v10.8.1.
Phage display selectionsA synthetic, phage-displayed Fab library42 was selected for binding to either integrin β2/integrin αM (R and D 4047-AM, antibodies 7062 and 7065) or integrin β2/integrin αL (R and D 3868-AV, antibodies 7060 and 7341) recombinant protein complexes. Briefly, integrin β2 recombinant protein complexes were immobilized on Maxisorp Immuno plates (Thermo Fisher, 12-565-135) and used for positive binding selections with library phage pools that were first exposed to neutravidin-coated wells to deplete nonspecific binders. After four rounds of binding selections, clonal phage was prepared and evaluated by phage ELISA and sequencing as previously described42.
Antibody productionAntibodies were produced using the human Expi293 expression system (Thermo Fisher). Expi293 cells (in a volume of 2 ml) were transiently transfected with construct DNA using FectoPro transfection reagent (Polyplus Transfection, 101000014). Following a 5-d expression period, antibodies were purified using rProteinA Sepharose (GE Healthcare) and stored in phosphate buffer (50 mM NaH2PO4, 75 mM Na2HPO4, 100 mM H3PO4 and 154 mM NaCl).
BLI binding assaysBinding of human integrin β2 antibodies was tested against three different integrin β2 complexes, including integrin β2/integrin αM (R and D 4047-AM), integrin β2/integrin αX (R and D 5755-AX) and integrin β2/integrin αL (R and D 3868-AV). To determine binding kinetic parameters, BLI was performed on an Octet HTX instrument (Sartorius) at 1,000 r.p.m. and 25 °C. All proteins were diluted in assay buffer (PBS, 1% BSA and 0.05% Tween 20). Tested and negative-control antibodies at 2 µg ml–1 were first captured on AHQ biosensors to achieve binding signals of 0.8–1.3 nm. Unoccupied Fc-binding sites on the antibody-coated sensors were subsequently quenched with 20 µg ml–1 Fc protein. After equilibration with assay buffer, biosensors were dipped for 600 s into wells containing a fivefold serial dilution of integrin β2 complexes (association phase), followed by transfer back into assay buffer for an additional 600 s (dissociation phase). Assay buffer alone served as a negative control. Binding response data were reference subtracted and globally fitted with a 1:1 binding model using ForteBio’s Octet Systems software v9.0.
Nonspecific ELISA panelThe ELISA protocol to assess interactions between the antibodies and unrelated macromolecules was performed as described previously56. The tested antigens included cardiolipin (50 μg ml–1; Sigma, C0563), keyhole limpet hemocyanin (5 μg ml–1; Sigma, H8283), lipopolysaccharide (10 μg ml–1; InvivoGen, tlrl-eblps), single-stranded DNA (1 μg ml–1; Sigma, D8899), double-stranded DNA (1 μg ml–1; Sigma, D4522) and insulin (5 μg ml–1; Sigma, I9278). In addition, binding of each antibody was also tested against empty wells (BSA-only control) and wells containing goat anti-human Fc (positive control, 1 µg ml–1; Jackson, 109-005-098). Antigens were coated at 30 µl per well in 384-well Maxisorp plates and incubated at 4 °C overnight. Plates were blocked with 0.5% BSA for 1 h at room temperature and washed with PBS + 0.05% Tween 20. Antibodies were added at 100 nM and allowed to bind for 60 min at room temperature. Plates were washed with PBS + 0.05% Tween 20, and binding was detected with anti-κ-horseradish peroxidase (1:5,000; Southern Biotech, 2060-05) and developed with TMB substrate (KPL (Mandel), KP-50-76-03).
Lentiviral construct generation and productionSecond-generation lentivirus constructs were used for transducing CAR expression cassettes in human T cells. Packaging plasmids used were pCMV delta R8.2 and pMD2.G. The transfer vector carrying the CAR expression cassette was pHR lentiviral vector with the SFFV promoter.
All transfer lentiviral plasmid constructs were generated using NEBuilder HiFi DNA Assembly master mix (New England Biolabs, E2621L) as per the vendor’s instructions with minor modifications. The DNA fragments containing the binder (scFv) sequence along with the 40-base pair vector-compatible flanking region for Gibson assembly were procured from Twist Bioscience. Target CAR plasmid backbone was linearized with BamHI-HF (New England Biolabs, R3136T). Stbl3 competent Escherichia coli cells (QB3 MacroLab, University of California, Berkeley) were used for transformation, and colonies obtained were screened by Sanger sequencing (Genewiz).
Lenti-X cells cultured in DMEM and plated 1 d before in a six-well plate (1 million cells per well) were transfected with the lentiviral plasmid constructs using polyethylenimine (Polysciences, 40,000 Da molecular weight, 24765-100). The spent media in cultures containing the lentivirus were collected 72 h after transfection. Twelve microliters of the stock concentration of polyethylenimine (2.5 mg ml–1) was used for transfecting 3 µg of plasmid per well. In total, 1.35 µg, 0.165 µg and 1.5 µg of lentiviral plasmids pCMV delta R8.2, pMD2.G and pHR (transfer vector), respectively, were used per well.
Human primary T cell isolationPrimary T cells were isolated from LeukoPaks (Stem Cell Technologies, 200-0092). CD8+ and CD4+ cells were isolated separately using an EasySep human CD8+/CD4+ T cell isolation kit (StemCell, 17952 (CD4); StemCell, 17953 (CD8)) based on magnetic bead separation. Isolated cells were stored frozen with 10% DMSO (MP Biomedicals, 196055). In total, primary T cells from five different donors were used for in vitro and in vivo studies.
Human CAR T cell generationT cells were thawed and grown in T cell medium consisting of Optmizer CTS medium (Gibco, A10221-01) + CTS supplement (Gibco, A10484-02) + 5% human AB serum (Valley Biomedical, HP1022) + penicillin/streptomycin + GlutaMAX (Gibco, 35050-061). Recombinant human IL-7 (Peprotech, 200-07) and IL-15 (Peprotech, 200-15; a final concentration of 10 ng ml–1 each) were freshly added to cells every 2–3 d. For manufacturing CAR T cells, primary T cells (CD4+ or CD8+) were thawed and cultured overnight. For aITGB2 CAR T cells, the cells were then additionally nucleofected with ribonuclease complex of ITGB2 sgRNA and Cas9 using a P3 Primary Cell 4D-Nucleofector X kit S (Lonza, V4XP-3032) and 4D-Nucleofector (Lonza) with its built-in program EO-115. Cells were then stimulated with 20 µl of CD3/CD28 Dynabeads (Thermo Fisher Scientific, 11131-D) per 1 million cells on day 0. The following day, lentivirus carrying the CAR expression cassette was added to the cells. The virus was withdrawn from the culture after 24 h, followed by two to three rounds of PBS washes by centrifugation. Dynabeads were magnetically withdrawn on day 4. On day 6 or 7, CAR+ cells were sorted by magnetic-activated cell sorting using the Myc tag of the CAR constructs with biotinylated c-Myc antibody (Milteni Biotec, 130-124-877). CAR T cells were used for studies within days 10–14 of culture.
CAR T cell cytokine analysisCAR T cells were cocultured with target (tumor) cells at a 1:1 E:T ratio for 24 h, and supernatant was snap-frozen in liquid nitrogen. Samples were analyzed at Eve Technologies Corporation with a Luminex 200 system (Luminex) using a Human High Sensitivity 14-Plex Discovery Assay (MilliporeSigma) according to the manufacturer’s protocol.
Retrovirus production and transductionHEK293T cells (3.5 × 106) were seeded in a 10-cm dish. Medium was replaced with 5 ml of complete DMEM (DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine), and cells were transfected with 7.5 μg of pCL-ECO plasmid and 7.5 μg of MSCV plasmid using Lipofectamine LTX with Plus reagent (Invitrogen, 15338030). The transfection mix was prepared in 3 ml of Opti-MEM medium (Gibco, 31985062) and incubated for at least 30 min at room temperature before being added dropwise onto the cell culture. Twenty-four hours after transfection, the medium was exchanged for 6 ml of complete DMEM collection medium. Retrovirus was collected, sterile filtered and frozen at −80 °C for storage at 24 and 48 h.
Mouse T cell isolation and cultureSpleens from mice were crushed and strained, and T cells were isolated using an EasySep mouse T cell isolation kit (StemCell Technologies, 19851). Cells were cultured in RPMI-1640 (Gibco, 11875093) supplemented with FBS (10%), penicillin–streptomycin (100 U ml–1), sodium pyruvate (1 mM), HEPES (10 mM), β-mercaptoethanol (Gibco, 21985-023), MEM non-essential amino acids (1×; Gibco, 11140050) and human IL-2 (200 U ml–1; Peprotech, 200-02).
Mouse CAR T cell generationThe scFv of the 7065 antibody or human CD19 scFv was cloned in the mouse CAR backbone (MSCV plasmid) with N-terminal mouse CD8a signal peptide and C-terminal mouse CD28–mouse CD3z domain, with P2A sequence followed by Thy1.1. Mouse T cells were cultured and activated for 24 h using Dynabeads Mouse T-Expander CD3/CD28 (Gibco, 11452D) and magnetically removed thereafter. T cells (2 × 106) were nucleofected with Cas9 ribonuclease complex (RNP; Lonza, V4SP-3096) using a 4D-Nucleofector 96-well unit (Lonza, AAF-1003S) and Lonza program code DN-100 with 60 pmol of Cas9 protein (QB3 MacroLab) and 120 pmol of sgRNA (UCCCUCCUCUAGAACUUCAC; Synthego) preincubated at 37 °C for 10–15 min. The culture medium was then added to the cells and incubated (37 °C, 5% CO2) for 1 h. Retronectin-coated (Takara, T100B) plates were used for culture, and 1 ml of retrovirus carrying the CAR expression cassette was added with 10 μg ml–1 polybrene. Cells were spinfected (2,000g, 30 °C, 60 min) and incubated overnight in a CO2 incubator at 37 °C. Medium was replenished regularly, and cell density was maintained at approximately 2 × 106 cells per ml.
T cell activation assayPBMCs were treated with 3 µM ionomycin (Sigma-Aldrich, 407950) + 25 ng ml–1 lipopolysaccharide (Sigma-Aldrich, L4391) + 100 U ml–1 IL-2 (Prospec, CYT-209) and cultured overnight. Cells were then co-stained with CD3 and CD69 and analyzed by flow cytometry.
In vitro cytotoxicity assayAML cell lines were engineered to stably express luciferase using lentiviral transduction. The cell lines were cocultured overnight with CAR T cells. d-Luciferin (150 μg ml–1; Gold Biotechnology, LUCK-1G) was added to each well and incubated for 3–5 min at room temperature, followed by luciferase detection using GloMax Explorer (Promega). For each ratio (CAR T cells:tumor cells), the bioluminescence readings from the tumor cells cocultured with untransduced T cells were considered 100% viable for normalization.
Degranulation assayCAR T cells were cocultured with tumor cells at ratio of 2:1 for 6 h at 37 °C with anti-CD107a and GolgiStop (BD Biosciences, 51-2092KZ). Cells were washed twice by centrifugation at 500g for 5 min at room temperature. Levels of CD107a were then measured with a flow cytometer as a readout of degranulation on GFP+ CAR T cells.
Generation of ITGB2-knockout cellsKnockout cell lines or primary T cells were generated using in vitro nucleofection of Cas9 ribonuclease protein complex. Briefly, 2 μl of each sgRNA (100 µM; Synthego Corporation) and recombinant Cas9 protein (40 µM; QB3 MacroLab, University of California, Berkeley) was incubated at 37 °C for 15 min to generate ribonuclease complex, which was then nucleofected using a 4D-Nucleofector (Lonza) with the built-in program DS-137 for cell lines (using Lonza V4XC-2032) and EO-115 for primary T cells (using Lonza V4XP-3032). The sgRNAs used in this study were obtained from the Brunello library57 (Supplementary Table 3).
Off-targeting analysis of sgRNAPotential off-target cleavage analysis was determined using the online tool CRISPOR58. The top five potential hits (genomic loci) were selected for PCR amplification (for primers, see Supplementary Table 2) from genomic DNA of ITGB2-knockout and wild-type primary human T cells extracted using a Monarch Genomic DNA Purification kit (New England Biolabs, T3010S). PCR amplicons were sequenced, and data were analyzed using the Synthego ICE Analysis Tool to determine the percentages of insertions and deletions.
Clonogenic assayCD34+ cells (1 × 103) from healthy donor GM-CSF-mobilized peripheral blood were co-incubated with aITGB2 CAR T cells, My96 CAR T cells, empty CAR T cells, untransduced T cells or medium only (IMDM, 2% FBS and penicillin/streptomycin) at an E:T ratio of 1:1 for 5 h in V-bottom 96-well plates in triplicate in methylcellulose-based medium supplemented with recombinant cytokines (MethoCult H4434 Classic, StemCell Technologies). After 13–14 d, colonies were classified and counted as granulocytes, erythrocytes, monocytes, megakaryocytes; granulocytes, monocytes; granulocytes; monocytes or erythrocytes. Images were acquired with a Keyence microscope using ×10, brightfield and color mode.
Generation of pathogen-specific T cellsCD45RA– healthy donor PBMCs were isolated using MACS MicroBead Technology (Miltenyi, 130-045-901) and pulsed with a pool of overlapping peptide libraries (15 mers overlapping by 11 amino acids) spanning the entire protein sequences of EBNA1, LMP1 and LMP2; BZLF1 and BRLF1 of Epstein–Barr virus; IE and pp65 of cytomegalovirus; hexon and penton of adenovirus and large T and VP1 or BK virus (JPT technologies) and expanded with IL-7 (10 ng ml–1) and IL-15 (5 ng ml–1; R&D Systems, 207-IL-010 and BT-015-010). On day 9, the cells received a second stimulation with irradiated pepmix-pulsed autologous ATCs pulsed with the same peptide libraries and irradiated HLA– lymphoblastoid cell lines (ULCLs) at a EBVST:ATC:K562cs/ULCL ratio (EBVST, Epstein-Barr virus-specific T cells; ATCs, activated T cells) of 1:1:5 with IL-7 and IL-15. Expanding cells were split as required and cryopreserved on day 16 of culture. Aliquots were thawed for analysis.
Generation of HIS miceThe NSG-SGM3 strain (NOD.Cg-PrkdcscidIl2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ) obtained from Jackson Laboratories was used to generate HIS mice. All mice used were 6–8 weeks old (either all male or all female for a particular study). Each mouse was treated with busulfan (12.5 mg per kg (body weight)) for 2 d consecutively, followed by 1 d of recovery and injection of 7 × 105 CD34+ human hematopoietic cells intravenously through the tail vein. Fully deidentified human CD34+-cell enriched blood samples were obtained from the Bone Marrow and Transplantation Laboratory at UCSF and were sorted by magnetic-activated cell sorting using a CD34 MicroBead kit (Miltenyi Biotec, 130-046-702) and incubated with anti-CD3 (Biolegend, 317302, clone OKT3) for T cell depletion 10 min before injection. Flow cytometry for human CD45+ cells was performed on mouse blood samples drawn 8 weeks later to determine engraftment efficiency (>1.5% threshold).
In vivo CAR T cell efficacy in PDX models of AMLAll mice used in the experiments were 6–8 weeks old (either all male or all female for a particular study) and were obtained from either Jackson Laboratories (NSG-SMG3) or were bred in-house (NSG) at the Preclinical Therapeutics Core of UCSF. In total, 1 × 106 Nomo1 cells were injected in each mouse. For PDXs, 2 × 106 cells were injected in each mouse irradiated with 250 cGy 4–6 h before injection. In total, 5 × 106 CAR T cells at a 1:1 ratio of CD4+:CD8+ CAR T cells were injected 5 d after tumor injection. For Nomo1 cells, tumor burden was determined by bioluminescence imaging with a Xenogen In Vivo Imaging System (Caliper Life Sciences). For PDXs, flow cytometry analysis of blood draws and ultrasonography of spleen size were used as readouts for tumor burden. No animals were excluded from the analysis. For such disseminated tumor models, our institution does not specify an allowable maximal tumor burden.
Statistics and reproducibilityAll statistical analyses were performed using GraphPad Prism v.9 unless stated otherwise. The data are represented as mean ± s.e.m., and P values of <0.05 were considered statistically significant. No statistical method was used to predetermine sample size, but our sample sizes are similar to those reported in previous publications11,16,44. Data distribution was assumed to be normal, but this was not formally tested. No data were excluded from the analyses. Animals were randomized based on body weight before treatment. The Preclinical Core Facility staff was blinded to mouse treatment and relevant outcomes. The other investigators were not blinded to allocation during the experiments and outcome assessment.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)