
記住我
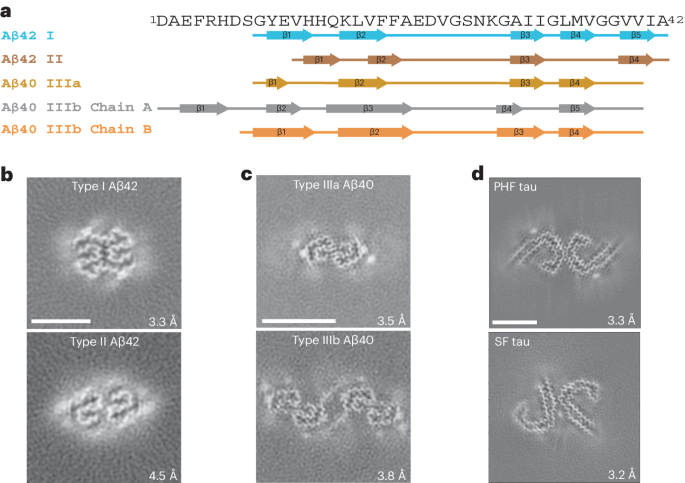

The human GR:Hsp90:FKBP52 complex was prepared by in vitro reconstitution of the complete GR chaperone cycle. GR DBD–LBD (residues 418–777 containing the F602S solubilizing mutation) (hereafter, GR) was incubated with Hsp70, Hsp40, Hop, Hsp90, p23 and FKBP52, allowing GR to progress through the chaperone cycle to reach the GR:Hsp90:FKBP52 complex (Extended Data Fig. 1a–c). The complex was stabilized with sodium molybdate, further purified and lightly crosslinked (Extended Data Fig. 1d,e). A 3.01 Å cryo-EM reconstruction of the GR:Hsp90:FKBP52 complex was obtained (Fig. 1a,b, Table 1 and Extended Data Figs. 1f and 2a,b), revealing a fully closed Hsp90 dimer (Hsp90A and Hsp90B) with a single GR and a single FKBP52, both occupying the same side of Hsp90 (Fig. 1a,b; for a discussion of Hsp90 nucleotide state, see Extended Data Fig. 3a and Methods). Despite using a multi-domain GR construct, only GRLBD was visible on the map.
Fig. 1: Architecture of the GR:Hsp90:FKBP52 complex.
a, Composite cryo-EM map of the GR:Hsp90:FKBP52 complex. Hsp90A, dark blue; Hsp90B, light blue; GR, yellow; FKBP52, teal. Color scheme is maintained throughout. b, Atomic model in cartoon representation with boxes corresponding to the interfaces shown in detail in c–g. c, Interface 1 of the Hsp90:GR interaction, depicting the Hsp90A Src loop (Hsp90A345–360) interacting with the GR hydrophobic patch. GR is in surface representation. d, Interface 2 of the Hsp90:GR interaction, depicting GR Helix 1 (GR532–539) packing against the entrance to the Hsp90 lumen. Hsp90A/Hsp90B are in surface representation. e, Interface 3 of the Hsp90:GR interaction, depicting GR pre-Helix 1 (GR519–531) threading through the Hsp90 lumen. Hsp90A/Hsp90B are in surface representation. f, Interface 1 of the Hsp90:FKBP52 interaction, depicting FKBP52 TPR H7e (FKBP52387–424) interacting with the Hsp90A/Hsp90B CTD dimer interface. Hsp90A/Hsp90B are in surface representation. g, Interface 2 of the Hsp90:FKBP52 interaction, depicting the Hsp90B MEEVD motif (Hsp90B700–706) binding in the helical bundle of the FKBP52 TPR domain. FKBP52 is in surface representation.
Hsp90 stabilizes GRLBD in a folded, ligand-bound stateIn the GR:Hsp90:FKBP52 complex, GRLBD is in a fully folded, ligand-bound conformation consistent with the conformation of the LBD in the GR–maturation complex (Extended Data Fig. 3b), but adopting a rotated position (discussed below). The folded GR is stabilized by Hsp90 at three major interfaces (Fig. 1c–e and Extended Data Fig. 3c–f): (1) Hsp90Src-loop:GRhydrophobic-patch, (2) Hsp90CTD:GRHelix1 and (3) Hsp90lumen:GRpre-Helix1 (for labeled structural motifs, see Extended Data Fig. 1c). In the first interface, Hsp90ASrc-loop (Hsp90345–360) flips out from the Hsp90lumen to interact with the previously described GRhydrophobic-patch13 (approximately 767 Å2 of buried surface area (BSA)) (Fig. 1c and Extended Data Fig. 3c). Along Hsp90ASrc-loop, Hsp90AF349,L351,F352,E353 contact GRHelix9/10 and the conserved, solvent-exposed Hsp90AW320 interacts with GRF774. Notably, Hsp90AW320,F349 also make contact with GR in the GR–loading complex and GR–maturation complex, although at quite different locations12,13. Additionally, there are multiple hydrogen bonds formed between Hsp90 N-terminal domain/middle domain (Hsp90NTD/MD) to GRHelix10 and GRK777.
Interface 2 is composed of Hsp90Y604 packing against GRHelix1 (GR532–539) and Hsp90Y627 sticking into a hydrophobic pocket formed by GRHelix3,4,9 (approximately 345 Å2 BSA) (Fig. 1d and Extended Data Fig. 3d,e), which was previously identified in the androgen receptor (AR) as a druggable hydrophobic pocket (BF3)40. In interface 3, the unstructured GRpre-Helix1 (GR519–531) is threaded through the Hsp90lumen (approximately 758 Å2 BSA)(Fig. 1e and Extended Data Fig. 3f). Two hydrophobic residues on GR (GRP522,P526) occupy two hydrophobic pockets within the Hsp90lumen. The interaction is further stabilized by multiple polar and hydrophobic interactions between GRpre-Helix1 and the Hsp90A/Hsp90B amphipathic helical hairpin (Hsp90606–628, Hsp90amphi-α) and Hsp90AMD/Hsp90BMD.
FKBP52 interacts with the closed Hsp90FKBP52 engages the closed Hsp90 at three major interfaces (Fig. 1f,g and Extended Data Fig. 4a–c): (1) FKBP52TPR/H7e:Hsp90ACTD/Hsp90BCTD, (2) FKBP52TPR:Hsp90BMEEVD and (3) FKBP52TPR:Hsp90BCTD. In interface 1, FKBP52H7e (FKBP52387–424) binds in a hydrophobic cleft formed by Hsp90ACTD/Hsp90BCTD at the closed dimer interface (approximately 1,109 Å2 BSA) (Fig. 1f, Extended Data Fig. 4a). Compared to the crystal structure, H7e breaks at positions FKBP52411–414 to allow hydrophobic residues (FKBP52L410,Y411,M414,F415,L418) to flip into the hydrophobic cleft formed by Hsp90CTD, consistent with the FKBP51H7e:Hsp90 interaction observed by cryo-EM19. Mutating the corresponding conserved residues on FKBP51H7e (FKBP51M412,F413 corresponding to FKBP52M414,F415) abolishes FKBP51:Hsp90 binding, indicating the importance of this binding site19. The interface is further stabilized by multiple hydrogen bonds and salt bridges from Hsp90ACTD/Hsp90BCTD to H7e flanking the helix break (Extended Data Fig. 4a). Furthermore, a portion of the Hsp90BMEEVD linker (Hsp90B700–706) binds along FKBP52H7e (Extended Data Fig. 4a). Helix 7e is found in many TPR-containing co-chaperones19; however, our structures, along with others, reveal the Helix 7e can bind Hsp90 in distinct positions due to sequence divergence41,42.
In interface 2, Hsp90BMEEVD binds in the FKBP52TPR helical bundle (approximately 779 Å2 BSA) (Fig. 1g and Extended Data Fig. 4b), with multiple hydrogen bonds, salt bridges and hydrophobic interactions, analogous to FKBP51:Hsp90MEEVD structures18,19. However, the MEEVD peptide binds in an opposite orientation relative to the FKBP52:Hsp90MEEVD crystal structure17, which may have been incorrectly modeled, as suggested18,43. Interface 3 is composed of FKBP52Helix5/6 in the TPR domain binding to Hsp90BCTD, stabilized by multiple hydrogen bonds (approximately 193 Å2 BSA) (Extended Data Fig. 4c), also observed in the FKBP51:Hsp90 cryo-EM structure19. While the interactions between FKBP52TPR/H7e:Hsp90 are conserved in the FKBP51:Hsp90 structure, the positions of the FKBP52 FK1 and FK2 domains are notably altered (Extended Data Fig. 4d), owing to the presence of GR, as discussed below.
FKBP52 directly binds GR, which is critical for GR functionUnexpectedly, FKBP52 directly and extensively interacts with GR, with all three FKBP52 domains wrapping around GR, cradling the folded, ligand-bound receptor near the GR ligand-binding pocket (Fig. 2a). The tertiary structure within each FKBP52 domain closely matches isolated domains from FKBP52 crystal structures; however, the interdomain angles are significantly different (Extended Data Fig. 4d), probably owing to the extensive interaction with GR. There are three major interfaces between FKBP52 and GR (Fig. 2b–d): (1) FKBP52FK1:GR, (2) FKBP52FK2:GR and (3) FKBP52FK2/TPR-linker:GRHelix12.
Fig. 2: The GR:FKBP52 interaction and functional significance.
a, Atomic model depicting the three interfaces between GR (yellow) and FKBP52 (teal) in the GR:Hsp90:FKBP52 complex. The FKBP52 proline-rich loop and PPIase catalytic site are highlighted in gray. Dexamethasone is colored in pink. b, Interface 1 between GR (yellow) and the FKBP52 FK1 domain (teal), showing interacting side chains and hydrogen bonds (dashed pink lines). c, Interface 2 between GR (yellow) and the FKBP52 FK2 domain (teal), showing interacting side chains and hydrogen bonds (dashed pink lines). d, Interface 3 between GR (yellow) and the FKBP52 FK2–TPR linker (teal), showing interacting side chains and hydrogen bonds (dashed pink lines). e, GR activation assay in wild-type yeast strain JJ762 expressing FKBP52 (‘52’) or FKBP52 mutants. The fold increase in GR activities compared to the empty vector (e.v.) control are shown (mean ± s.d.). n = 3 biologically independent samples per condition. Significance was evaluated using a one-way analysis of variance (F(5,12) = 26.10; P < 0.0001) with post-hoc Dunnett’s multiple comparisons test (n.s. P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). P values: P(e.v. versus 52) <0.0001, P(52 versus 52ΔFK1) <0.0001, P(52 versus 52 S118A) <0.0001, P(52 versus 52 Y161D) 0.0003, P(52 versus 52 W259D) 0.0005. f, Sequence alignment of eukaryotic FKBP52 showing conserved residues involved in the GR:FKBP52 interaction (denoted by a black asterisk). The bottom aligned sequence is human FKBP51. The alignment is colored according to the ClustalW convention. g, GR protein sequence conservation mapped onto the GR atomic model from the GR:Hsp90:FKBP52 complex. Residue conservation is depicted from most variable (cyan) to most conserved residues (maroon). GR residues that interact with FKBP52 are shown as spheres.
In interface 1, FKBP52FK1 interacts with a large surface on GR, canonically used for GR dimer formation, consisting of the post-Helix 1 strand (Helix 1–3 loop), Helix 5 and β1,2 (approximately 280 Å2 BSA) (Fig. 2b). Three-dimensional variability analysis in CryoSparc revealed that the interaction between FKBP52FK1 and GR is highly dynamic, even as the other FKBP52 domains (FK2 and TPR) remain stably associated with GR (Supplementary Movies 1 and 2). At the FK1:GR interface, GRY545 on the post-Helix 1 strand interacts with a hydrophobic surface formed by the FKBP5281–88 loop and forms a hydrogen bond with FKBP52Y113. Supporting this interaction, residues of GRpost-Helix1 (GR544–546) have previously been implicated in FKBP51/52-dependent regulation of GR activity44,45. In addition, the FKBP52 proline-rich loop (β4–β5 loop or 80s loop) contacts GRHelix5/β1,2. Three-dimensional variability analysis in CryoSparc revealed that the proline-rich loop positioning is flexible, deviating from the position in the crystal structure (Protein Data Bank (PDB) ID: 4LAV) (ref. 46) and adopting different interfaces with GR (Supplementary Movies 3 and 4). In the consensus 3D refinement map, the proline-rich loop adopts a position similar to the crystal structure, and FKBP52A116,S118,P119 interact with GRHelix5/β1,2. The FKBP52P119L mutation has been shown to reduce GR and AR activation in vivo, while FKBP52A116V has been shown to increase AR activation in vivo29. We also demonstrate that the FKBP52S118A mutation significantly reduces FKBP52-dependent GR potentiation in vivo (Fig. 2e), further demonstrating the functional significance of this interaction site. In addition, S118 has been identified as a phosphorylation site on FKBP52, but not FKBP51 (qPTM database47) (possibly due to the unique adjacent proline, FKBP52P119, which could recruit proline-directed kinases). Phosphorylation at FKBP52S118 may help promote the interaction between the proline-rich loop and GR, which could also explain the large effect of the FKBP52S118A mutation in vivo.
While FKBP52FK1 is known to have PPIase enzymatic activity, GR is not bound in the PPIase active site and, accordingly, no GR prolines were found to have been isomerized compared to other GR structures (PDB IDs: 1M2Z (ref. 48) and 7KRJ (ref. 13)). Consistent with this, mutation of GR prolines does not disrupt FKBP52-dependent regulation of GR45. Additionally, mutations that disrupt PPIase activity do not affect FKBP52-dependent GR potentiation in vivo29. Conversely, PPIase inhibitors have been shown to block the FKBP52-dependent potentiation of GR in vivo23. This can now be understood, as docking of PPIase inhibitors (FK506 and rapamycin) into the PPIase active site demonstrates that the inhibitors would sterically block the FKBP52FK1:GR interface (Extended Data Fig. 4e), as previously hypothesized23,29.
Interface 2 is composed of the FKBP52 FK2Y161 sticking into a shallow hydrophobic pocket formed by GRHelix3, GRHelix11–12 loop (GRT561, M565,E748), and a hydrogen bond between the FKBP52 backbone and GRE748 (approximately 125 Å2 BSA) (Fig. 2c). Supporting this interaction, we show that the FKBP52Y161D mutation significantly reduces FKBP52-dependent GR potentiation in vivo, demonstrating the importance of this interaction (Fig. 2e). In interface 3, the solvent exposed, conserved FKBP52W259 on the FK2–TPR linker makes electrostatic and hydrophobic interactions with GRHelix12 (approximately 235 Å2 BSA) (Fig. 2d), which adopts the canonical agonist-bound position even in the absence of a stabilizing co-activator peptide interaction48 (Extended Data Fig. 3b). We show that the corresponding FKBP52W259D mutation significantly reduces FKBP52-dependent GR potentiation in vivo, demonstrating the functional importance of this single residue (Fig. 2e). Interestingly, FKBP52W259 is also conserved in the FKBP-like co-chaperone XAP2 and a recent structure reveals XAP2 engages with an Hsp90–client using the analogous XAP2W168, suggesting this residue is critical more broadly for FKBP co-chaperone:client engagement42. At interface 3, FKBP52K254,E257,Y302,Y303 make further polar interactions between the FK2–TPR linker and GRHelix12 (Fig. 2d). While a significant portion of the GRHelix12 co-activator binding site is available in the FKBP52-bound GR, the N-terminus of a co-activator peptide would sterically clash with FKBP52TPR based on the GR:co-activator peptide structure48 (Extended Data Fig. 5b). Thus, co-activator binding in the nucleus could help release GR from its complex with Hsp90:FKBP52. We also find that the residues at the FKBP52:GR interfaces are conserved across metazoans (Fig. 2f,g) and have been identified as sites that crosslink to GR in vivo (FKBP52Y159, W257) (ref. 49), in agreement with our results that single point mutations at each of the three FKBP52:GR interfaces has a significant effect on GR function in vivo. Based on the observation that FKBP52TPR is sufficient to bind Hsp90 and Hsp90:SHR complexes19,20, the FKBP52 single point mutants probably do not disrupt FKBP52 binding to the GR:Hsp90 complex, but specifically disrupt the GR:FKBP52 interaction and prevent stabilization of GRLBD by FKBP52.
FKBP52 advances GR to the next stage of maturationWe previously described another GR:chaperone complex, the GR–maturation complex (GR:Hsp90:p23) (ref. 13), which also contains a closed Hsp90 dimer and a folded, ligand-bound GR (Fig. 3a). However, in the GR:Hsp90:FKBP52 complex, GR is rotated by approximately 45° relative to the GR–maturation complex (Fig. 3a). Hsp90ASrc-loop interacts with GRpre-Helix1 in the maturation complex, but flips out to stabilize the rotated GR position in the GR:Hsp90:FKBP52 complex by interacting with the GRhydrophobic-patch (Fig. 3b,c). In both complexes, GRpre-Helix1 is threaded through the Hsp90lumen; however, in the GR:Hsp90:FKBP52 complex, GR has translocated through the Hsp90lumen by two residues, positioning prolines (GRP522,P526) in the hydrophobic pockets of the lumen rather than leucines (GRL525,L528) (Fig. 3d). This translocation positions GRLBD further from Hsp90, probably allowing enough space for the observed GR rotation. Despite the translocation and rotation of GR, Hsp90 uses the same surfaces to bind GR (Hsp90Bamphi-α, Hsp90ASrc-loop and Hsp90AW320); however, the contact surfaces on GR are different. The rotation of GR may facilitate LBD dimerization, which is on pathway to activation. In the GR–maturation complex, dimerization of the LBD clashes with Hsp90CTD; however, due to the rotation of the LBD in the GR:Hsp90:FKBP52 complex, LBD dimerization would now be sterically permitted after FKBP52 release (Extended Data Fig. 5c).
Fig. 3: FKBP52 competes with p23 to bind GR:Hsp90.
a, Atomic model of the GR–maturation complex (top) and the GR:Hsp90:FKBP52 complex (bottom) with boxes corresponding to the interfaces shown in detail in b–d. Hsp90A, dark blue; Hsp90B, light blue; GR, yellow; p23, green; FKBP52, teal. b, Position of the Hsp90A Src loop in the GR–maturation complex (Hsp90A, cyan) versus the GR:Hsp90:FKBP52 complex (Hsp90A, dark blue). GR (yellow, surface representation). Hsp90A Src loop residues interacting with the GR hydrophobic patch are shown. c, Interface between the p23 tail-helix (green) and the GR hydrophobic patch (yellow, surface representation) in the GR–maturation complex (top). The p23 tail-helix is replaced by the Hsp90A Src loop (dark blue) in the GR:Hsp90:FKBP52 complex (bottom), which flips up to interact with the GR hydrophobic patch (yellow, surface representation). Interacting side chains are shown. d, Interaction between GR pre-Helix 1 (GR523–531) in the Hsp90 lumen in the GR–maturation complex (top) versus the GR:Hsp90:FKBP52 complex (bottom). Hsp90A/Hsp90B are in surface representation colored by hydrophobicity. GR pre-Helix 1 translocates through the Hsp90 lumen by two residues in the transition from the GR–maturation complex to the GR:Hsp90:FKBP52 complex. e, Equilibrium binding of 10 nM F-dex to 100 nM GR DBD–LBD with chaperones and 15 μM FKBP52 (‘52’) (mean ± s.d.). n = 3 biologically independent samples per condition. ‘Chaperones’: 15 μM Hsp70, Hsp90, Hop, and p23 or p23Δhelix; 2 μM Ydj1 and Bag-1. Significance was evaluated using a one-way analysis of variance (ANOVA) (F(3,8) = 541.2; P < 0.0001) with post-hoc Šídák’s test (n.s. P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001). P values: P(chaperones versus chaperones + 52) 0.0002, P(chaperones + 52 versus chaperones with p23Δhelix + 52) <0.0001, P(chaperones with p23Δhelix versus chaperones with p23Δhelix + 52) <0.0001. f, Equilibrium binding of 10 nM F-dex to 100 nM GR DBD–LBD with chaperones, 15 μM FKBP52 (‘52’) and 20 mM sodium molybdate (‘Mo.’) (mean ± s.d.). n = 3 biologically independent samples per condition. ‘Chaperones’: 15 μM Hsp70, Hsp90, Hop and p23; 2 μM Ydj1 and Bag-1. ‘-p23’ indicates p23 was left out of the chaperone mixture. Significance was evaluated using a one-way ANOVA (F(5,12) = 761.5; P < 0.0001) with post-hoc Šídák’s test (n.s. P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001). P value <0.0001 for each comparison.
FKBP52 competes with p23 through allosterySurprisingly, FKBP52 competes with p23 to bind the GR:Hsp90 complex, although there is no direct steric conflict between FKBP52 and p23 binding (Fig. 3a). During 3D classification on the cryo-EM dataset, GR:Hsp90:p23 complexes were observed at low abundance (~74,000 particles); however, the GR:Hsp90:FKBP52 complexes showed no apparent p23 density (Extended Data Fig. 2a,b), despite p23 being present at high concentration in the reconstitution. Furthermore, FKBP52 was only found associated with the rotated GR position, while the GR position in the p23-containing classes was only consistent with the GR–maturation complex. Thus, FKBP52 appears to specifically bind the rotated GR position, which is not compatible with p23 binding. This is consistent with mass spectrometry studies, demonstrating FKBP52 competes with p23 to form a stable GR:Hsp90:FKBP52 complex50. In the rotated GR position, Hsp90ASrc-loop flips out of the Hsp90lumen to bind the GRhydrophobic-patch, which was previously engaged by the p23tail-helix (Fig. 3a–c). Thus, rotation of GR dictates the accessibility of the GRhydrophobic-patch to either Hsp90 or p23. FKBP52 stabilizes the rotated position of GR and therefore favors GR binding to Hsp90ASrc-loop over p23.
FKBP52 potentiates GR ligand binding in vitroTo quantitatively assess the functional significance of FKBP52 on GR activation, we added FKBP52 to the in vitro reconstituted GR chaperone cycle, using the GR DBD–LBD construct (residues 418–777 containing the F602S solubilizing mutation) and monitored GR ligand binding, as previously described11,13. Addition of FKBP52 to the GR chaperone cycle resulted in the enhancement of GR ligand binding above the already enhanced GR + chaperones control reaction at equilibrium (Fig. 3e), strongly suggesting FKBP52 potentiates the GR ligand binding affinity beyond the minimal chaperone mixture, consistent with reports in vivo23. We hypothesized that FKBP52 functions in a similar manner to the p23tail-helix in stabilizing the ligand-bound GR. As previously described, removal of the p23tail-helix (p23Δhelix) resulted in a decrease in GR ligand binding activity in the GR chaperone system13; however, addition of FKBP52 to the reaction fully rescued GR ligand binding in the p23Δhelix background (Fig. 3e). Thus, FKBP52 can functionally replace the p23tail-helix, probably by also directly stabilizing the ligand-bound GR. Given that FKBP52 and the p23tail-helix bind different locations on GR, the mechanisms of stabilization may be unique. Additionally, in the p23Δhelix background, FKBP52 potentiated ligand binding to a greater extent than in the wild-type p23 background. We hypothesize that removing p23tail-helix alleviates the competition between p23 and FKBP52, allowing p23 to remain bound to the GR:Hsp90:FKBP52 complex. Given that p23 is known to stabilize the closed Hsp90 conformation13,51, the enhanced ligand binding in the p23Δhelix background may be due to stabilization of closed Hsp90 by p23Δhelix. Interestingly, FKBP52 also affected GR ligand binding independent of Hsp90, with addition of FKBP52 to GR resulting in enhanced ligand binding, probably due to an Hsp90-independent chaperoning effect15,52 (Extended Data Fig. 5d).
Upon Hsp90 closure, FKBP52 can functionally replace p23Given that FKBP52 can functionally replace p23tail-helix, we wondered whether FKBP52 could also functionally replace p23 altogether. p23 is known to stabilize Hsp90NTD closure through the globular p23 domain13,51 in addition to stabilizing the ligand-bound GR through the p23tail-helix13. Omitting p23 from the GR chaperone cycle drastically reduces GR ligand binding, as previously described11,13. The addition of FKBP52 in place of p23 results in a small increase in ligand binding but does not fully rescue ligand binding activity (Fig. 3f). We reasoned this could be due to the inability of FKBP52 to sufficiently stabilize Hsp90 closure, as previously suggested53. Therefore, we added molybdate to these reactions, which stabilizes Hsp90NTD closure by acting as a γ-phosphate analog in the Hsp90NTD ATP-binding site13,54. Addition of molybdate to the reaction lacking p23 resulted in a small increase in GR ligand binding but did not fully rescue ligand binding activity. However, addition of molybdate to the reactions containing FKBP52 without p23 resulted in a full reactivation of ligand binding and even potentiated ligand binding over the control GR + chaperones reaction (Fig. 3f), much like with p23Δhelix. Thus, FKBP52 is able to functionally replace p23 if Hsp90NTD closure is stabilized. Taken together, these results suggest FKBP52 can stabilize the ligand-bound GR, like p23, but cannot stabilize the closed Hsp90NTD conformation, which requires p23.
GR:Hsp90:FKBP51 structure determinationIn vivo, the interplay between FKBP52 and the highly similar FKBP51 have profound implications for GR activity. FKBP51 is functionally antagonistic to FKBP52-dependent potentiation of GR in vivo; thus, the relative ratios of FKBP51 and FKBP52 dictate GR activity levels23,28,55. To understand how FKBP51 antagonizes FKBP52, we prepared the human GR:Hsp90:FKBP51 complex by in vitro reconstitution of the GR chaperone cycle with FKBP51 (Extended Data Fig. 1d–f). We obtained a 3.23 Å cryo-EM reconstruction of GR:Hsp90:FKBP51 (Fig. 4a,b, Table 1 and Extended Data Fig. 6a,b). Contrary to our expectations, the FKBP51-containing structure appears nearly identical to the FKBP52-containing structure. The GR:Hsp90:FKBP51 structure reveals a fully closed Hsp90 dimer complexed with a single GR and a single FKBP51, which occupy the same side of Hsp90 (Fig. 4a,b; for a discussion of Hsp90 nucleotide, see Extended Data Fig. 7a and Methods). As with the FKBP52 dataset, GR:Hsp90:p23 complexes were also observed during 3D classification and the GR:Hsp90:FKBP51 complexes showed no apparent p23 density (Extended Data Fig. 6a,b). The FKBP51:Hsp90 interactions are analogous to the FKBP52:Hsp90 interactions, including Hsp90BMEEVD:FKBPTPR and Hsp90CTD:FKBPH7e, also seen in the Hsp90:FKBP
留言 (0)