Clinical history and neuropathology
Human tissue samples were from the Dementia Laboratory Brain Library at Indiana University School of Medicine. Their use in this study was approved by the ethical review processes at the institution. Informed consent was obtained from patients’ next of kin.
We studied two individuals with Down syndrome. Case 1 (DS-1) was a 59-year-old male who died with neuropathologically confirmed diagnosis of AD after a 6 year history of progressive dementia. He also had a clinical history of a seizure disorder with multifocal myoclonic jerks. A brain autopsy was carried out. The fresh brain weighed 750 g with severe atrophy in the frontal and temporal lobes. Moderate to severe neuronal loss and gliosis were seen in the frontal cortex, temporal cortex, parietal cortex, hippocampus, thalamus, midbrain, pons, medulla and cerebellum. Numerous plaques and neurofibrillary tangles were present in these same areas. The AD neuropathologic change was ranked as high AD neuropathologic change with Thal phase 5 (A3), Braak stage VI (B3) and Consortium to Establish a Registry for Alzheimer’s Disease score of C3 (A3, B3 and C3). Moderate CAA was present. Case 2 (DS-2) was a 46-year-old male who died with neuropathologically confirmed diagnosis of AD after over a 6 year history of progressive dementia. Both his mother and maternal grandmother had a history of dementia. A brain autopsy was carried out. The fresh brain weighed 1,056 g. There was mild symmetric atrophy, with mild to moderate atrophy of the left cerebral hemisphere. The left and right hemibrains weighed 526 and 516 g, respectively. Mild atheromatous change was present in the middle cerebral arteries. Mild to moderate numbers of neuritic plaques were observed in the neocortex, amygdala, hippocampus, entorhinal cortex and midbrain. NFTs, neuronal loss, gliosis and moderate to severe CAA were present. The AD neuropathologic change was ranked as intermediate AD neuropathologic change with Thal phase 3 (A2), Braak stage IV (B2) and Consortium to Establish a Registry for Alzheimer’s Disease score of C2 (A3, B2 and C2). Tissue samples for neuropathological studies were obtained from representative brain regions. The 8 μm thick brain sections were used. For immunohistochemistry, primary antibodies were 4G8 (Abcam, 1:1,000), β-amyloid 1–40 (Millipore Sigma, 1:400), β-amyloid 1–42 (Millipore Sigma, 1:400) and AT8 (anti-phospho tau, Thermo Fisher, 1:300). The signal from the antibodies was visualized using avidin–biotin followed by horseradish peroxidase-conjugated streptavidin and the chromogen diaminobenzidine. Immunohistochemical sections were counterstained with hematoxylin.
Genetics
To confirm trisomy 21, we performed chromosomal microarray analysis on genomic DNA extracted from brain using a whole genome platform that includes both nonpolymorphic and single-nucleotide polymorphism oligonucleotide probes (Affymetrix CytoScan HD Microarray). Patient hybridization results were compared to data pooled from hundreds of normal individuals. To assess for common copy number variations (CNVs) in the populations and regions of clinical significance, databases potentially consulted include, but are not limited to, the International Standards for Cytogenomic Arrays Clinical CNV Database, Database of Genomic Variants, DECIPHER Population CNV Database, Online Mendelian Inheritance in Man, ClinGen, ClinVar and PubMed. All results are analyzed and reported using the February 2009 National Center for Biotechnology Information human genome build 37.1 (hg19).
Filament extraction
Sarkosyl-insoluble fractions were prepared from freshly frozen frontal cortex of DS-1 and temporal cortex of DS-2, as previously described36,42. Briefly, ~2 g of tissue was homogenized in 20 volumes (w/v) extraction buffer consisting of 10 mM Tris–HCl, pH 7.4, 0.8 M NaCl, 1 mM EGTA and 10% sucrose. Samples were centrifuged at 20,000g and the supernatants were brought to 2% sarkosyl and incubated at 37 °C for 1 h. Samples were centrifuged at 10,000g for 10 min. The supernatants were spun at 100,000g for 1 h at 4 °C. The resulting pellets were resuspended in 1 ml g−1 tissue in the extraction buffer and centrifugated at 3,000g for 5 min. This supernatant was further purified by threefold dilution in buffer consisting of 50 mM Tris–HCl, pH 7.5, 0.15 M NaCl, 10% sucrose and 0.2% sarkosyl, followed by centrifugation at 100,000g for 30 min at 4 °C. The final pellet was resuspended in 20 mM Tris–HCl, pH 7.5 and 50 mM NaCl and stored at 4 °C.
Western blotting
Samples were sonicated for 1 min, boiled with gel 2× Laemmli sample buffer (Bio-Rad) for 5 min at 100 °C and resolved on 4–12% Bis-Tris gels for Aβ-amyloid or 10% Bis-Tris gels for tau (NuPAGE). For HFIP treatment, samples were centrifuged at 200,000g for 30 min and HFIP added to the pellet, sonicated and left overnight at 37 °C. Samples were dried under nitrogen, washed three times with water and centrifuged at 200,000g for 30 min. The final pellet was resuspended in loading buffer and resolved on 4–12% Bis-Tris gels. Proteins were transferred to nitrocellulose membranes and the membranes were incubated with blocking solution (5% nonfat milk in phosphate-buffered saline with 0.1% Tween 20). Membranes were incubated for 1 h with primary antibody diluted in TBS. Antibodies used were 4G8 (BioLegend, 1:1,000), 6E10 (BioLegend, 1:1,000), AT8 (Thermo Fisher, 1:1,000) and HT7 (Thermo Fisher, 1:1,000). After incubation with secondary antibody for 45 min, proteins were visualized using a chemiluminescence kit (SuperSignal West Pico, ThermoFisher) according to the manufacturer’s specifications.
Immuno-EM
For immuno-EM, samples were analyzed as previously described28,36. AT8 antibody or D5452 antibody (anti-amyloid-β, Cell Signaling) were diluted 1:50 in 0.1% gelatin in phosphate-buffered saline and incubated overnight at 4 °C. Secondary antibodies used were 6 nm anti-mouse and 10 nm anti-rabbit immunogold particles (Electron Microscopy Sciences). Negative staining was performed with NanoVan (Ted Pella) for 5 s at room temperature. Images were taken on a Tecnai G2 Spirit Twin scope equipped with an AMT CCD Camera.
Mass spectrometry sample preparation
Samples were diluted in 8 M urea, 50 mM Tris–HCl pH 8.5 (100 µl), reduced with 5 mM Tris(2-carboxyethyl)phosphine hydrochloride for 30 min at 37 °C and alkylated with 10 mM chloroacetamide at room temperature in the dark, for 30 min. Samples were digested in two steps with LysC/trypsin (Promega). After overnight trypsin digestion in 2 M urea, the samples were applied to Pierce detergent removal spin columns (Thermo Scientific) and then desalted on SepPak 18 cartridge (Waters Corporation) washed with 1 ml of 0.1% trifluoroacetic acid, eluted in 600 µl of 70% acetonitrile/0.1% formic acid (FA) and dried by speed vac.
Liquid chromatography with tandem mass spectrometry
Samples were reconstituted in 50 µl of 0.1% FA and 7 µl were injected on an Easynano LC1200 coupled with Aurora 25 cm column (IonOpticks) insonation column oven (40 °C) on an Eclipse Orbitrap mass spectrometer (Thermo Fisher Scientific). Peptides were eluted on a 115 min gradient from 5% to 35% B, increasing to 95% B over 10 min and decreasing to 5% B for 5 min (solvent A: 0.1% FA; solvent B: 80% acetonitrile, 0.1% FA). The instrument was operated with FAIMS pro 4 coefficients of variation (−30, −45, −55 and −65 V), positive mode, 0.6 s cycle time per coefficient of variation with APD and Easy-IC on. Full scan included 400–1,500 m/z with 60,000 resolution, standard automatic gain control and auto max IT, 40% RF lens, 5 × 104 intensity threshold, charge states 2–8 and 30 s dynamic exclusion with common settings. MS2 parameters of 1.6 m/z quadrupole isolation, 30% fixed higher-energy collision dissociation cell, 15,000 Orbitrap resolution, standard automatic gain control and dynamic IT were included.
Mass spectrometry data analysis
Raw files were loaded into PEAKS X Pro Studio 10.6 Build 20201221 (Bioinformatics Solutions). The precursor ion tolerance was 10 ppm 0.02 Da. Peptides obtained after trypsin digestion were used for database searches of the reviewed Uniprot_Swissprot Homo sapiens database and common contaminants (20,437 entries) with variable post-translational modifications (PTMs). PEAKS PTM and SPIDER searches were enabled to search all de novo peptides above a 15% score for over 300 potential PTMs and mutations. A 0.1% peptide false discovery rate cutoff (−10 log P ≥21.8), PTM A score >10, mutation ion intensity >1 and de novo only score >80% were applied to the data, followed by PEAKS LFQ analysis. Raw and searched data are available at ProteomeXchange. The bioinformatic analysis Gene Ontology of identified proteins was done by DAVID Bioinformatics Resources 6.8 (refs. 43,44). P value was represented as −log10. The Venn diagram was generated using BioVenn45.
High‑resolution cryo‑EM imaging
Cryo-EM grids of brain extracts of the two patients with DS were prepared in a biosafety level 2 cabinet while wearing appropriate personal protective equipment. A total of 2–3 µl of the sample were applied on a graphene oxide-coated EM grid, then washed with 10 mM Tris pH 7.8 before vitrifying using a semi-automated Gatan CP3 cryo-plunger. High-resolution cryo-EM movies were collected on a FEI Titan Krios at 300 kV with a Gatan K3 detector mounted on a quantum energy filter with 20 eV slit width (Tables 1 and 2). For DS-1, we recorded 12,540 movies of 50 frames per movie with an exposure time of 48 ms per frame, with a dose rate of 21 electrons per Å2 per frame for a total accumulated dose of 51.45 electrons per Å2 at a pixel size of 1.054 Å. For DS-2, we recorded 24,881 movies of 50 frames per movie with an exposure time of 52 ms per frame and a dose rate of 1.054 electrons per Å2 per frame for a total accumulated dose of 52.35 electrons per Å2 at a pixel size of 1.054 Å. The datasets were collected with defocus values ranging from −0.5 to −1.5 μm. The movies were gain corrected, motion corrected and dose weighted using MotionCor2 (ref. 46). The contrast transfer function of all aligned and non-dose-weighted micrographs was estimated using CTFFIND-4.1 (ref. 47).
Helical reconstruction
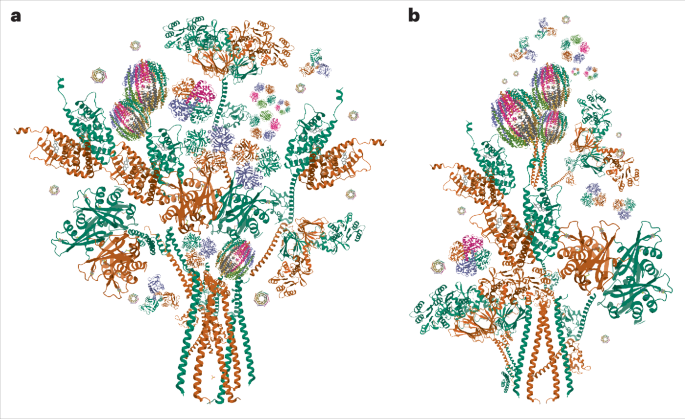
Image processing was performed in RELION 4.0 (ref. 48). Filaments were picked manually for DS-1 and automatically for DS-2 using RELION helical picker as end-to-end line segments. The initial quantity of particles for DS-2 was excessively high from the automatic over picking to ensure that all filaments were picked. We extracted all the helical segments with a box size of 600 pixels (632 Å), downscaled to 200 pixels to speed up analysis, and an inter-box distance of ~15 Å. Several rounds of reference-free 2D classifications were carried out to remove nonfilament contaminants and to find homogeneous subsets using a regularization value of T = 1–2. Aβ and tau filaments were visually identified from the 2D class averages. For both, a new 256 pixel box size, without downscaling, was used to re-extract the filament segments with inter-box distance of approximately 15 Å. The initial 3D reference maps were reconstructed de novo from best 2D class averages comprising a full helical crossover using relion_helix_inimodel2d. The initial round of 3D classification was low-pass filtered to 10 Å. Several rounds of 3D classification were carried out to obtain the best homogeneous subset. The final selected segments were used for final 3D auto-refinement with optimization of the helical twist and rise to yield a 3D map showing clearly visible β-strand separation and side-chain densities. For the less abundant type III filaments, we first reconstructed the type IIIb filaments consisting of a dimer of dimer packing of the protofilaments. For type IIIa filaments, which have a pitch similar to that of type IIIb filaments but are narrower, we assumed that the type IIIa filaments corresponded to one of the two dimers in the type IIIb filaments. A dimer (for example, half of the type IIIb filament) was segmented, centered and examined using HI3D49, which showed that the two protofilaments in the dimers are packed with a 21 screw symmetry instead of C2 symmetry. These analyses helped obtain the initial model and helical parameters to reconstruct the type IIIa filament structure. Bayesian polishing was subsequently applied, followed by contrast transfer function refinement. A 3D classification was done to remove suboptimal segments, along with another round of 3D auto-refinement with optimization of the helical twist and rise. We used a 10–30% z percentage to generate the mask for post-processing and resolution estimation. The final reconstructions were sharpened using the standard post-processing procedures in RELION. The overall resolution was calculated from Fourier shell correlations at 0.143 between two independently refined half-maps, employing phase randomization for the convolution effects correction of an optimized, soft-edged solvent mask as implemented in the trueFSC.py program in JSPR software (Extended Data Fig. 8)50.
Atomic modeling and structural analysis
The previously deposited model was fitted into the sharpened density maps using ChimeraX51. The central chain of each model was manually adjusted in Coot52. The atomic positions of all models were refined with their respective helical symmetry parameters using Rosetta53. The identity and the sequence range of the modeled proteins were validated using the map2seq Web app54. Final atomic models were validated using MolProbity55 (Tables 1 and 2). All models’ figures were generated in ChimeraX51 (Extended Data Fig. 9).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.

Comments (0)