
Remember me



Unless noted otherwise, all chemicals were purchased from Carl Roth, Thermo Fisher Scientific, Sigma-Aldrich or Tokyo Chemical Industry and were used directly without further purification. Gases were purchased from Air Liquide.
Molecular cloningAll used primers were purchased from Eurofins Genomics and are listed in Supplementary Table 1. Polymerase chain reactions (PCRs) were conducted with Q5 High-Fidelity DNA Polymerase (New England Biolabs), PCR purifications were conducted with the Monarch PCR & DNA Cleanup Kit (New England Biolabs), extraction of genomic DNA was conducted with the Monarch Genomic DNA Purification Kit (New England Biolabs), Gibson assemblies were conducted with the NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs), and Golden Gate cloning was conducted with the NEBridge Golden Gate Assembly Kit (New England Biolabs) according to the instructions provided by the manufacturer. Successful assembly of desired vectors was verified by Sanger sequencing through Microsynth Seqlab. All plasmids used and created in this study are listed in Supplementary Table 2.
The pK18mobSacB knockout plasmids were generated via Golden Gate cloning or Gibson assembly. For Gibson assembly, backbone amplification was always done with the primers oMM0227 and oMM0228. The upstream and downstream homologous regions of the targeted genomic loci were amplified from R. capsulatus B10S genomic DNA with primers featuring overhangs suitable for Golden Gate cloning or Gibson assembly. The primers used for the construction of each knockout plasmid are listed in Supplementary Table 1. The plasmid pBS85-BsaI-genR, used for the interruption of the anfHDGK locus by a gentamycin resistance cassette, was constructed via Golden Gate cloning. First, a BsaI cutting site was introduced into pBS85 using the primers oMM0027 and oMM0028 to create pBS85-BsaI. Next, Golden Gate inserts were amplified. The gentamycin resistance cassette was amplified from pOGG024 using oMM0033 together with oMM0034. In parallel, the upstream and downstream homologous regions of the anfHDGK locus were amplified from R. capsulatus B10S genomic DNA using oMM0035–oMM0038. Eventually, the three inserts and the pBS85-BsaI plasmid were combined in a Golden Gate reaction to generate pBS85-BsaI-genR.
For the generation of pOGG024-kanR, a kanamycin resistance cassette was amplified via PCR from plasmid pRhon5Hi-2 using primers oMM0384 and oMM385. The plasmid pOGG024 was linearized via PCR with oMM0386 and oMM0387. The two DNA amplicons were purified and combined via Gibson assembly, which yielded pOGG024-kanR. The construction of the pOGG024-kanR-anfHDGK expression plasmid was achieved in four steps. First, the anfHDGK operon was amplified from R. capsulatus B10S genomic DNA via PCR using the primers oMM0021 and oMM0146. In parallel, the destination plasmid pRhon5Hi-2 was linearized with oMM0145 and oMM0023. Following purification of the PCR products, pRhon5Hi-2-anfHDGK was generated via Gibson assembly. Next, two BsaI cutting sites were removed via a modified version of QuikChange mutagenesis56 using the primers oMM0161–oMM0164. The resulting plasmid pRhon5Hi-2-anfHDGK is suitable for Golden Gate cloning and was used as a template to amplify the anfHDGK expression cassette with the primers oMM0389 and oMM0390. The resulting PCR product was purified and subsequently inserted into pOGG024-kanR via Golden Gate cloning. Lastly, affinity tags (Strep-tag II and His6-tag) for protein purification were inserted via restriction-free cloning57 using primers oMM0223 and oMM0224 for the insertion of the Strep-tag II at the AnfD C terminus and oMM0510 and oMM0511 for the insertion of the His6-tag at the AnfH N terminus. Both tags were inserted without any linker sequence. Eventually, the sequence of the pOGG024-kanR-anfHDGK expression plasmid was confirmed by whole-plasmid sequencing through Plasmidsaurus.
Genetic manipulation of R. capsulatusStarting from the wild-type strain B10S, the R. capsulatus genome was successively modified to generate an ideal strain for the recombinant expression and subsequent purification of the Fe nitrogenase. For the deletion of anfHDGK, a gentamycin resistance cassette was inserted into the anfHDGK locus, thereby interrupting the operon (Extended Data Fig. 1). The plasmid pBS85-BsaI-genR was introduced into R. capsulatus B10S via conjugational transfer as described in ref. 33, selecting for the gentamycin resistance conferred by the transferred vector. Subsequently, double-recombinant clones were identified through screening for gentamycin resistance and tetracycline sensitivity (resulting from the loss of the suicide vector backbone through double recombination) on peptone yeast agar plates33 containing 15 µg ml−1 gentamycin or 10 µg ml−1 tetracycline, respectively. Positive clones were further investigated via colony PCR to check the anfHDGK locus. The purified PCR products were analyzed by Sanger sequencing to identify clones with a successfully modified anfHDGK operon. Building up on the ΔanfHDGK::genR mutant of R. capsulatus B10S, all further deletions were achieved successively via the sacB scarless deletion method described in ref. 58. In brief, sequences of around 500 base pairs homologous to the upstream and downstream regions flanking the gene of interest were generated and cloned into a pK18mobSacB suicide vector (see above). The resulting plasmid was conjugated into the R. capsulatus recipient strain33, selecting for the kanamycin resistance conferred by the suicide vector. Intermediate strains derived from single colonies that were obtained from the previous step were passaged three times in liquid peptone yeast medium33, growing each passage for 24 h at 30 °C and moderate shaking in the dark. The final passage was spread on a peptone yeast agar plate containing 5% (m/V) sucrose, which was then incubated for 72 h at 30 °C under an Ar atmosphere and illumination by six 60-W krypton lamps (Osram Licht). Single colonies of R. capsulatus growing on the sucrose-containing agar plate were screened for kanamycin and sucrose sensitivity on peptone yeast plates containing 50 µg ml−1 kanamycin or 5% (m/V) sucrose, respectively. Colonies that could tolerate sucrose but were not growing on kanamycin-containing agar plates were further investigated via colony PCR to check the targeted genomic locus. Lastly, the purified PCR products were analyzed by Sanger sequencing (Microsynth Seqlab) to identify successful knockout clones. Genomic DNA of the modified R. capsulatus B10S strain (MM0425) was extracted and sequenced via next-generation sequencing (Novogene) to confirm the deletions listed in Table 1. The R. capsulatus MM0436 expression strain was generated by introducing the pOGG024-kanR-anfHDGK expression plasmid into MM0425 via conjugational transfer. All used strains are listed in Supplementary Table 3.
Growth medium and conditions for protein productionRhodobacter capsulatus was cultivated phototrophically at 32 °C under a 100% N2 atmosphere. Cultivation on agar plates was conducted on peptone yeast agar plates33 selective for the respective expression plasmid. Liquid cultures of R. capsulatus were cultivated diazotrophically in a modified version of RCV medium33 that contained 30 mM DL-malic acid, 0.8 mM MgSO4, 0.7 mM CaCl2, 0.05 mM sodium ethylenediaminetetraacetic acid (Na2EDTA), 0.03 mM thiamine hydrochloric acid, 9.4 mM K2HPO4, 11.6 mM KH2PO4, 5 mM serine, 1 mM iron(III) citrate, 45 µM B(OH)3, 9.5 µM MnSO4, 0.85 µM ZnSO4, 0.15 µM Cu(NO3)2 and 25 µg ml−1 kanamycin sulfate at a pH set to 6.8. For protein production, the expression strain was inoculated from a glycerol stock on peptone yeast agar plates and cultivated for 48 h. The obtained cell mass was used to inoculate liquid cultures in N2-flushed RCV medium, which were cultivated for 24 h. Subsequently, 800 ml of RCV medium were inoculated with an optical density at 660 nm (OD660) of 0.1 for protein production. Protein purification was initiated when the cultures reached an OD660 of ~3.0.
Protein purificationAll protein purification steps were carried out strictly anaerobically under a 95% Ar and 5% H2 atmosphere inside a COY tent (Coy Laboratory Products). All buffers were anaerobized by flushing them with Ar and equilibrating them for at least 12 h inside the COY tent before use. For collection, sodium dithionite was added to a final concentration of 5 mM to each liquid culture, which were then centrifuged at 15,970 × g for 60 min at 10 °C. The liquid supernatant was decanted, and the cell pellets were resuspended and combined in high-salt buffer (50 mM Tris (pH 7.8), 500 mM NaCl, 10% glycerol, 4 mM sodium dithionite) supplemented with 0.2 mg ml−1 bovine pancreatic deoxyribonuclease I and one cOmplete EDTA-free Protease Inhibitor Tablet (Roche). Subsequently, cells were disrupted by three passages through a French Press cell disruptor (Thermo Fisher Scientific, FA-078AE) at 20,000 p.s.i. The obtained lysate was centrifuged at 150,000 × g for 60 min at 8 °C, and the liquid supernatant was filtered (pore size, 0.2 µm). The entire cleared cell extract was then applied sequentially to high-salt buffer equilibrated HisTrap HP (Cytiva) and Strep-Tactin XT 4Flow high-capacity (IBA Lifesciences) columns via an ӒKTA pure chromatography system (Cytiva). After extensive washing with high-salt buffer, the catalytic component was eluted from the Strep-Tactin XT column with high-salt buffer supplemented with 50 mM biotin. Fractions containing the catalytic component were pooled, the buffer was exchanged with low-salt buffer (50 mM Tris (pH 7.8), 150 mM NaCl, 10% glycerol, 4 mM sodium dithionite) with a Sephadex G-25 packed PD-10 desalting column (Cytiva) and the protein was concentrated with an Amicon Ultra-15 Centrifugal Filter Unit (molecular weight cutoff, 100 kDa; Merck Millipore). Meanwhile, the HisTrap HP column was washed extensively with high-salt buffer containing 25 mM imidazole before eluting the reductase component with high-salt buffer plus 250 mM imidazole. The eluate was directly subjected to SEC on a HiLoad 26/600 Superdex 200 pg column (Cytiva) equilibrated with low-salt buffer. AnfH2 eluted in a single peak at around 205 ml and was subsequently concentrated using an Amicon Ultra-15 Centrifugal Filter Unit (molecular weight cutoff, 30 kDa; Merck Millipore). Protein yields for both nitrogenase component fractions were determined using the Quick Start Bradford 1x Dye Reagent (Bio-Rad Laboratories) according to the instructions provided by the manufacturer, and the purity of both protein fractions was analyzed via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Following the described protein purification procedure, approximately 3.5 mg of catalytic component and 12 mg of reductase component were obtained per liter of R. capsulatus growth culture. Eventually, both nitrogenase components were flash frozen and stored in liquid N2 until further use.
SDS–PAGE analysisFor SDS–PAGE, protein samples were denatured by boiling them with Pierce Lane Marker Reducing Sample Buffer (Thermo Fisher Scientific) for 10 min at 98 °C. After centrifuging the samples at 17,000 × g, the clear supernatant was loaded on a 4–20% Mini-PROTEAN TGX Stain-Free Gel (Bio-Rad Laboratories) including PageRuler Plus Prestained Protein Ladder (Thermo Fisher Scientific) as a molecular weight reference. The electrophoresis was run for 30 min at a constant voltage of 180 V before staining the gel with GelCode Blue Safe Protein Stain (Thermo Fisher Scientific).
Nitrogenase turnover assaysNitrogenase activity was assessed in vitro by measuring specific activities for H2 and NH3 formation under an N2 or Ar atmosphere. Working under an Ar atmosphere, varying amounts of AnfH2 were dissolved in an anaerobic solution of 50 mM Tris (pH 7.8), 10 mM sodium dithionite, 3.5 mM ATP, 7.87 mM MgCl2, 44.59 mM creatine phosphate and 0.20 mg ml−1 creatine phosphokinase (Sigma-Aldrich, C3755). The reaction vials were sealed by crimping them with butyl rubber stoppers, and the headspace was exchanged to N2 or Ar. Next, the reactions were initialized by adding 0.1 mg of Anf(DGK)2 up to a total volume of 700 µl and were allowed to proceed for 8 min at 30 °C with moderate shaking at 250 r.p.m. Reactions were quenched with 300 µl of 400 mM Na2EDTA solution (pH 8.0), and the amounts of formed H2 and NH3 were analyzed as described below.
Quantification of H2Amounts of formed H2 were determined via headspace analysis using a Clarus 690 GC system (gas chromatography with a flame ionization detector and a thermal conductivity detector, GC–FID/TCD; PerkinElmer) with a custom-made column circuit (ARNL6743) that was operated with TotalChrom v6.3.4 software (PerkinElmer). The headspace samples were injected by a TurboMatrixX110 autosampler (PerkinElmer), heating the samples for 15 min to 45 °C prior to injection. The samples were then separated on a HayeSep column (7’ HayeSep N 1/8’ Sf; PerkinElmer), followed by molecular sieve (9’ Molecular Sieve 13×1/8’ Sf; PerkinElmer) kept at 60 °C. Subsequently, the gases were detected with an FID (at 250 °C) and a TCD (at 200 °C). The quantification of H2 was based on a linear standard curve that was derived from measuring varying amounts of H2 under identical conditions. The results were plotted using GraphPad Prism v9 software (Dotmatics).
Quantification of NH3Quantification of in vitro generated NH3 was done with a modified version of a fluorescence NH3 quantification method described in ref. 59. One hundred microliters of sample were combined with 1 ml of a solution containing 2 mM o-phthalaldehyde, 10% (V/V) ethanol, 0.05% (V/V) β-mercaptoethanol and 0.18 M potassium phosphate buffer (pH 7.3) and incubated for 2 h at 25 °C in the dark. Fifty microliters of each sample were transferred into individual wells of a black Nunc F96 MicroWell plate (Thermo Fisher Scientific), and fluorescence at 485 nm was monitored with an Infinite 200 PRO plate reader (Tecan) in fluorescence top reading mode using an excitation wavelength of 405 nm. The quantification of NH3 was based on a linear standard curve that was derived from measuring varying amounts of NH4Cl under identical conditions. Samples incubated under an Ar atmosphere instead of N2 were used to correct for background signal. The results were plotted using GraphPad Prism v9.
Mass photometryMass photometry measurements were carried out on microscope coverslips (1.5 H, 24 × 50 mm; Carl Roth) with CultureWell Reusable Gaskets (CW-50R-1.0, 50–3 mm diameter × 1 mm depth) that had been washed with three consecutive rinsing steps of distilled H2O and 100% isopropanol and dried under a stream of pressurized air. Measurements were set up in gaskets assembled on microscope coverslips on the stage of a TwoMP mass photometer (Refeyn) with immersion oil. Samples were measured in anaerobic measurement buffer (150 mM NaCl, 50 mM Tris (pH 7.8), 10% glycerol, 10 mM sodium dithionite) after focusing on the glass surface using the droplet-dilution focusing method. After focusing, 0.5 µl of nitrogenase sample (500 nM stock concentration, dissolved in measurement buffer with 4 mM dithionite) were removed from an anaerobic vial, quickly added to 19.5 µl of measurement buffer, and mixed on the stage of the mass photometer. Measurements were started ~5 s after removing protein from the anaerobic environment. Data were acquired for 60 s at 100 frames per second using AcquireMP v2.3 (Refeyn). Mass photometry contrast was calibrated to molecular masses using 50 nM of a mixture of citrate synthase complexes with varying complex stoichiometries of masses ranging from 86 kDa to 430 kDa. Mass photometry data sets were processed and analyzed using DiscoverMP v20222 R1 (Refeyn). The details of mass photometry image analysis have been described previously60.
Metal analysisMetal analysis was done using ICP-OES. For sample preparation, 0.12 mg and 0.24 mg of catalytic and reductase components, respectively, were dissolved in 0.5 ml of trace metal grade concentrated nitric acid and incubated for 12 h at 25 °C. Subsequently, the samples were boiled for 2 h at 90 °C before they were diluted 17-fold in distilled water. The metal content was analyzed with a 720/725 ICP-OES device (Agilent Technologies) on iron (λ = 238.204 nm), molybdenum (λ = 202.032 nm), nickel (λ = 216.555 nm) and zinc (λ = 213.857 nm). The device was operated with ICP Expert v4.1.0 software (Agilent Technologies). All analyzed metals were quantified using ICP multi-element standard solution IV (Merck) as a standard. The results were plotted using GraphPad Prism v9.
Preparation of AlF3-stabilized nitrogenase complexStabilized Fe nitrogenase complex consisting of two reductase components and one catalytic component was prepared as described in ref. 61. In brief, 4 nmol of catalytic component and 32 nmol of reductase component were combined in 100 mM MOPS, 50 mM Tris, 100 mM NaCl (pH 7.3) with 5 mM sodium dithionite, 4 mM NaF, 0.2 mM AlCl3, 8 mM MgCl2 and 1 mM ATP in a total volume of 4 ml. The reactions were incubated for 1 h at 30 °C before they were concentrated with an Amicon Ultra-0.5 ml Centrifugal Filter Unit (molecular weight cutoff, 100 kDa; Merck Millipore). Subsequently, less than 500 µl of sample were injected via the ӒKTA pure chromatography system onto a Superdex 30 Increase 10/300 GL column (Cytiva) equilibrated with 50 mM Tris (pH 7.8), 200 mM NaCl and 5 mM sodium dithionite. Elution fractions from the peak corresponding to the appropriate molecular weight species (expected molecular weight of catalytic component combined with two reductase components is ~372 kDa) were pooled, and the presence of all nitrogenase subunits was confirmed via SDS–PAGE as described above.
Cryo-EM sample preparation and data collectionFour microliters of protein solution (total protein concentration, 1 mg ml−1) were applied to freshly glow-discharged QUANTIFOIL R2/1 300 copper mesh grids (Quantifoil Micro Tools) and blotted for 5 s with a blot force of 5 at ~90% humidity and 8 °C using a Vitrobot Mark IV (Thermo Fisher Scientific) that was placed inside an anaerobic COY tent. For CHAPSO detergent-supplemented grids, 1 µl of detergent (dissolved in the same buffer as the protein) was added to a final concentration of 0.4% (m/V) to 3 µl of protein solution on the respective grid. Grids were plunge frozen in a liquid ethane (37 vol%) and propane (63 vol%) mix and stored in liquid nitrogen until data collection. CHAPSO-supplemented grids of AnfDGK were prepared to prevent preferential orientation.
Data of cryo-EM samples were collected on a Titan Krios G3i electron microscope (Thermo Fisher Scientific), operated at an acceleration voltage of 300 kV and equipped with a BioQuantum K3 energy filter (Gatan). Data were collected in electron counting mode at a nominal magnification of ×105,000 (0.837 Å per pixel) with a total dose of 50 e−/Å2 (50 fractions), using the aberration-free image-shift correction in EPU v2.9–2.11 software (Thermo Fisher Scientific). The nominal defocus range used for data collection was −1.4 μm to −2.4 μm.
Cryo-EM data processingAll data sets were processed entirely in cryoSPARC v4.1 (ref. 62). For all data sets, dose-fractionated movies were gain-normalized, aligned, and dose-weighted using Patch Motion correction, and the contrast transfer function (CTF) was determined using the Patch CTF routine. The information regarding cryo-EM data collection, model refinement and statistics are listed in Table 2.
Processing the AnfHDGK–AlF3 complexBlob picker and manual inspection of particles were used to extract an initial 2,114,475 particles with a box size of 300 pixels, which were used to build two-dimensional (2D) classes. 2D classes with protein-like features were used to initialize template picking. After manual inspection and extraction with a box size of 300 pixels, this yielded a total of 3,365,366 particles, which were used to build 2D classes. After selecting 2D classes with protein-like features, the selected particles were used to train a model that was subsequently used to pick particles using Topaz (ref. 63). A total of 1,706,699 candidate particles were extracted with a box size of 380 pixels and cleaned from non-particle candidates by 2D classification into 200 classes. Selected particles were used for ab initio reconstruction and classification into four classes. Particles of the two best-aligning classes (432,216 particles) were subjected to further cleaning by three-dimensional (3D) classification into ten classes with a target resolution of 5 Å. 3D classification yielded volumes containing zero, one or two AnfG subunits, with unchanged orientation of the remaining subunits. The best-aligning classes with one or more AnfG subunit bound (218,653 particles) were subjected to local CTF refinement, local motion correction, and subsequent non-uniform refinement with C2 symmetry, two extra final passes, 15-Å initial low-pass resolution, 12-Å gold-standard Fourier shell correlation (GSFSC) split resolution, 4-Å dynamic mask near expansion, 10-Å dynamic mask far expansion, 8-Å dynamic mask start resolution, per-particle defocus optimization, and Ewald sphere (EWS) correction, yielding a global resolution of 2.35 Å and a temperature factor of −76.7 Å2. Further classification did not yield improved resolution.
Processing the AnfDGK componentInitial attempts to solve the AnfDGK complex structure without reductase component used grids prepared without detergent (CHAPSO). Standard processing workflows of this data set (blob picking, template picking, Topaz picking and manual picking) yielded 2D classes that exclusively showed one orientation (Extended Data Fig. 5a). Resulting ab initio and 3D reconstructions failed to yield initial volumes with a nitrogenase-like shape. We therefore focused our efforts on grids prepared in the presence of 0.5% CHAPSO.
Here, blob picker and manual inspection of particles were used to extract an initial 2,018,560 particles with a box size of 320 pixels from 2,000 micrographs, which were used to build 2D classes. 2D classes with protein-like features were used to train a Topaz model to pick particles, which was subsequently used to re-extract particles from the same 2,000 micrographs for downstream 2D classification and Topaz model training. A total of 1,647,264 particles were extracted with a box size of 340 pixles and cleaned from non-particle candidates by 2D classification. Cleaned particles were used to train a Topaz model on 4,578 micrographs and subsequently used to pick particles from all 18,320 micrographs. A total of 7,962,489 particles were extracted with a box size of 324 pixels and cleaned by three subsequent rounds of 2D classification into 200, 100 and 50 classes, respectively (Extended Data Fig. 5b). Selected particles of the last 2D classification step (2,121,950) were used for ab initio reconstruction and classification into four classes. Particles of the two best-aligning classes (1,336,362 particles) were subjected to further cleaning by 3D classification into ten 3D classes with a target resolution of 4 Å. 3D classification yielded no volumes containing electron density at positions where AnfG would be expected. Nevertheless, particles of the three best-aligning classes (304,619 particles) were used for non-uniform refinement with C1 symmetry and no additional corrections. This yielded a 2.64-Å global resolution map that contained no indication of electron density at locations where AnfG would be expected, nor at select regions of AnfDK in close contact with AnfDK. A subsequent non-uniform refinement using particles of the seven best-aligning classes (563,245) from the 3D classification, the 2.64-Å map as an input volume, C2 symmetry, CTF correction, defocus correction and EWS correction yielded a map with a global resolution of 2.49 Å. This map also contained no electron density at locations where AnfG would be expected, nor at regions in AnfDK that would be near the expected AnfG position. Further classification was not attempted, as AnfG could not be detected in processed volumes.
Model building and refinementInitial cryo-EM map fitting was performed in UCSF Chimera v1.16 (ref. 64) using AlphaFold 2 (ref. 39) models for AnfD, AnfK and AnfG, as well as an AnfH crystal structure (PDB 7QQA) from A. vinelandii40. The resulting model was manually built further in Coot v0.8.9.2 (ref. 65). Automatic refinement of the structure was done using phenix.real_space_refine of the Phenix v1.21.1 software suite66. Manual refinements and water picking were performed with Coot v0.8.9.2. The FeFeco was built with REEL of the Phenix software suite. The model statistics are listed in Table 2.
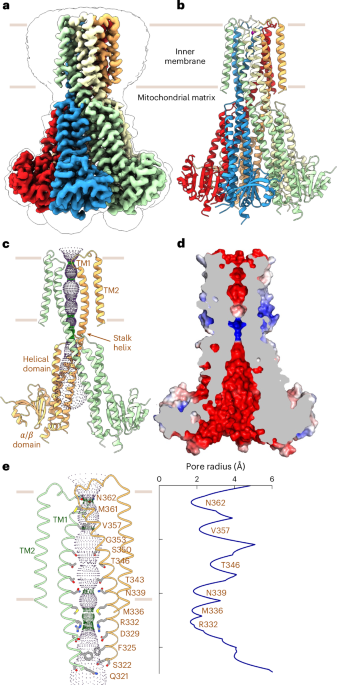
Substrate channel calculationSubstrate channels were calculated using CAVER v3.0.3 software (ref. 49). The coordinates of sulfur atom S2B were provided as the starting point for channel calculations. The probe radius, shell radius and shell depth were set to 0.7, 4.0 and 5.0 Å, respectively. Many channels were predicted using CAVER v3.0.3. However, the two most probable channels with the shortest length, the largest bottleneck radius, the highest throughput and prioritization by CAVER v3.0.3 were selected and are displayed throughout the manuscript as surfaces generated in PyMOL v2.5 (Fig. 3d).
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)