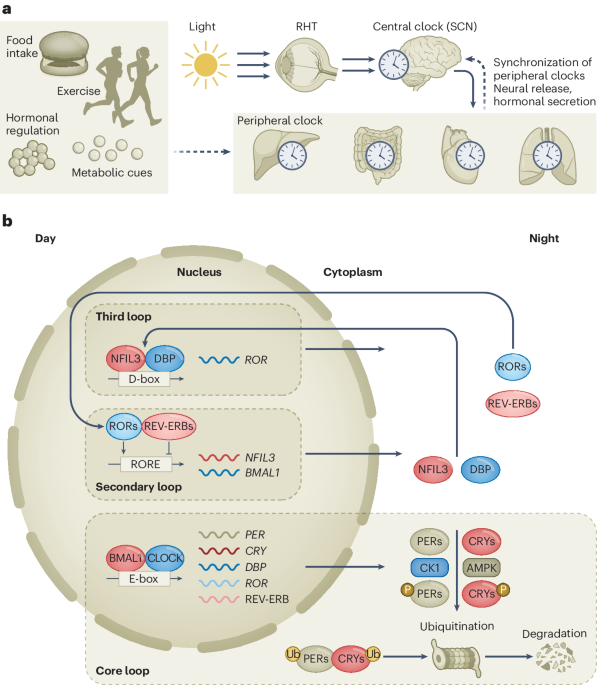
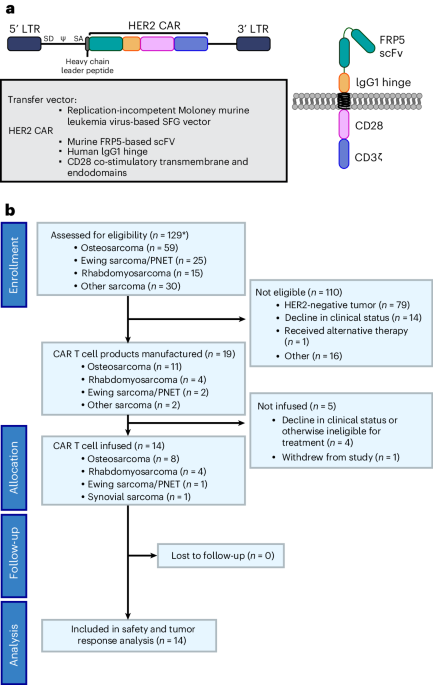
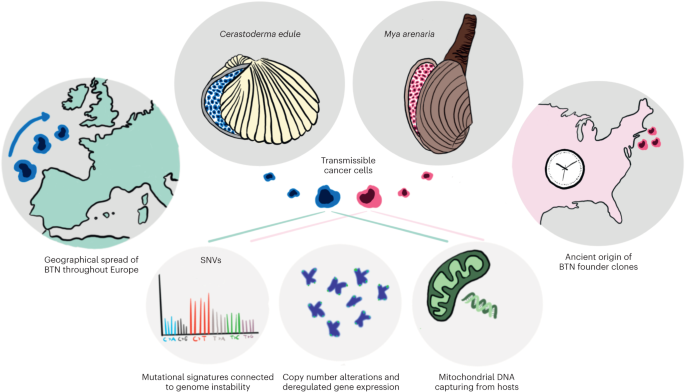
The experiments in this study were performed in compliance with the Institutional Animal Care and Use Committee (IACUC) protocol no. IA16-00507; the institutional review board (IRB) of the University of New York Grossman School of Medicine (no. S17-00651) was used for the PDX-related experiments.
Cell culture
PaTu-8988T, PANC1, MiaPaCa2 and PaTu-8902 cells were obtained from ATCC or the Deutsche Sammlung von Mikroorganismen und Zellkulturen. Primary murine PDAC cell lines (HY19636 and HY15549) were isolated from tumors from C57BL/6 (B6) genetically engineered mice (LSL-KrasG12D; p53L/+, Ptf1aCre+) as described previously46. All cell lines were grown in 5% CO2 and 37 °C. All cell lines were cultured in DMEM (catalog no. 10-017-CV, Corning) with 10% FCS (catalog no. S11550H, Atlanta Biologicals) and 1% penicillin-streptomycin (catalog no. 15140122, Thermo Fisher Scientific). Cells were tested routinely for Mycoplasma contamination using PCR. Cell lines were authenticated by periodic fingerprinting and visual inspection; low-passage cultures were carefully maintained in a central laboratory cell bank.
Chemicals
13C5-labeled Gln (catalog no. CLM-1822-H), alpha-15N Gln (catalog no. NLM-1016-PK), 13C6-labeled glucose (catalog no. CLM-1396-PK), 15N-labeled alanine (catalog no. NLM-454) and 13Cx,15Nx-labeled amino acid standard mix (catalog no. MSK-A2) were acquired from Cambridge Isotope Laboratories. N-acetyl-l-cysteine (catalog no. A7250) dimethylsulfoxide (DMSO), methoxyamine hydrochloride and MTBSTFA + 1% t-BDMSC and DON (catalog no. D2141) were obtained from Sigma-Aldrich. Trametinib (catalog no. 16292) and dacomitinib (pan-RTK inhibitor, catalog no.9001879) were obtained from Cayman Chemical.
Cell proliferation and viability assays
Proliferation assays were performed at various densities depending on the growth kinetics of each line28. HY19636 and HY15549 were plated at 1,000 cells per well; MiaPaCa2, PANC1 and PaTu-8988T were plated at 2,000 cells per well in a 96-well plate. Cells were allowed to attach overnight. The next day, cells were treated with DON at increasing concentrations, as indicated in the figure legends. Cell growth was assessed every 12 h using Cytation C10 (Agilent Technologies) for 72 h. Confluency was derived from the object sum area divided by the total area. Every cell proliferation assay was performed with at least two technical replicates and biological replicates. For the Gln rescue assay, cells were plated at 1,000 cells per well and supplemented with the indicated Gln concentration (10 mM, 4 mM, 2 mM, 0.5 mM). DON was added the next day; cellular confluency was obtained after 7 days using Cytation C10.
OCR
Basal OCR was measured using a Seahorse XFe96 analyzer (Agilent Technologies) as described previously47. PDAC lines were plated at 10,000–20,000 cells per well depending on the growth kinetics of the line in either standard DMEM or with DON for 24 h before the assay. Before the assay, the medium was removed and replaced with the XF assay medium (25 mM glucose, 4 mM Gln, no sodium bicarbonate, 5 mM HEPES); the plate was incubated for approximately 45 min at 37 °C in a non-CO2 incubator (Thermo Fisher Scientific). Medium was changed before the beginning of the assay; drug treatments were maintained in the XF assay medium throughout the experiment. Cell numbers were normalized to protein quantification after the assay using the DC Protein Assay Kit (Bio-Rad Laboratories). At least two independent experiments were performed for all conditions; data from 6–12 technical replicate wells for each separate experiment were averaged. Data were plotted relative to each independent group’s untreated or control condition.
Metabolomics
For metabolite extraction, cells were washed with 0.9% NaCl prepared in high-performance liquid chromatography-grade water to remove medium and other contaminants. As described previously, cellular metabolites were collected using a methanol-water-chloroform solution47,48. Cellular metabolites were vortexed at 4 °C for 15 min. To separate the inorganic layers, samples were centrifuged at maximum speed for 10 min. Polar metabolites were obtained from the aqueous phase and transferred (300 µl) into polypropylene vials (catalog no. 5190-2243, Agilent Technologies) and dried down by SpeedVac (Thermo Savant, catalog no. SPD111V, Thermo Fisher Scientific). Once metabolites were dried, 20 µl methoxyamine hydrochloride (20 mg ml−1 in pyridine, always prepared fresh) was added and incubated for 60 min at 37 °C. Next, 20 µl MTBSTFA + 1% t-BDMSC was added and incubated for 30 min at 37 °C. Samples were analyzed using GC–MS using a gas chromatograph (7890B model, Agilent Technologies) with a DB-35ms Ultra Inert column (catalog no. 122-3832UI, Agilent Technologies) installed and associated with an Agilent 5977B mass spectrometer. The GC–MS parameters, quantification and correction for natural isotope abundances were performed as described previously28,47,48. For the 13C6-glucose and 13C5-Gln tracing experiments,15N-Gln or 15N-alanine cells were plated in a six-well plate at 2.0 × 105 cells per well and allowed to attach overnight in DMEM. Next, cells were washed with PBS twice and cultured for 24 h in DMEM supplemented with 4 mM 13C5-Gln, 25 mM 13C6-Glucose or 1 mM 15N-Alanine; 10% dialyzed serum was added. For the tracing studies, cells were pretreated with DON (25 µM) overnight, medium was removed and washed with PBS. Then, cells were incubated with fresh DON and with stable isotopes for 24 h. For amino acid and metabolite profiling, metabolites were collected using an 80% methanol solution with 1 µg norvaline and labeled with amino acid standards (catalog no. MSK-A2-1.2, Cambridge Isotope Laboratories) as described elsewhere28,47,48. For the LC–MS analysis, cells were treated for 24 h with PBS or DON. Samples were subsequently processed by the Metabolomics Core Research Laboratory at New York University (NYU) Langone Health, as described previously47.
Mice
All animal studies were approved by the NYULMC IACUC (protocol no. IA16-00507). Female B6J mice (C57BL/6J, Taconic), athymic (NCrNU, Taconic) or NSG (The Jackson Laboratory) used in this study were 8–10 weeks old and not involved in previous procedures. All mice were maintained in the animal facility of the NYU Grossman School of Medicine with access to a standard diet (PicoLab Rodent diet 20, catalog no. 5053) and water ad libitum at constant ambient temperature and a 12-h light cycle. Animal numbers were determined according to previous work28,49. Maximum tumor burden was limited to less than 2 cm in any direction; this metric was not exceeded in this study.
Orthotopic xenograft experiments
Orthotopic injections of PDAC cells were conducted as described previously28. Briefly, mice were anesthetized with ketamine (120 mg kg−1) and xylazine (10 mg kg−1) before surgery. An incision was made in proximity to the spleen and the pancreas was carefully externalized. Cells were resuspended in 20 µl of a solution consisting of Hank’s balanced salt solution (HBSS) and Matrigel (catalog no. 356231, Corning) at a 1:1 ratio and injected into the tail of the pancreas using insulin syringes (29-gauge needle, catalog no. 324702, BD). For HY19636 and HY15549 cells, approximately 40,000 cells were injected unless indicated otherwise. For PANC1, 50,000 cells were injected into nude mice. After surgery, the incision was closed using a 3-0 coated VICRYL VIOLET suture (catalog no. J311H, Ethicon) and using the BD AutoClip Wound Closing System. Mice were treated with buprenex every 12 h after surgery for 48 h. Animals were allowed to recover from surgery for at least 7 days before starting the experimental process. Animals were randomly assigned before starting the experimental process, unless indicated otherwise. Experimental treatment started 7 days after surgery. Tumor weight was measured at the end point after mice were euthanized and tumor was collected for further analysis (for example, histology, bulk RNA-seq).
As described previously49, for the hemi-splenic injections, 1 × 105 cells were resuspended in 100 µl HBSS and incorporated into an insulin syringe (28-gauge needle, catalog no. 329461, BD). The spleen was separated using ligating clips (catalog no. 002200, Teleflex), and cells were injected into the hemi-spleen. After injection, the splenic vein was ligated with ligating clips (catalog no. 001200, Teleflex) at the hilum of the spleen; then the hemi-spleen was removed. The peritoneum was closed with a 3-0 VICRYL VIOLET suture and the skin was closed using the BD AutoClip Wound Closing System.
For the DRP-104 studies, mice were treated daily either with vehicle (Tween-80:Ethanol:Saline 5:5:90 v/v/v) or with DRP-104 (3 mg kg−1) intraperitoneally for two cycles (5 days on, 2 days off) unless indicated otherwise. DRP-104 was a gift from Dracen Pharmaceuticals. DRP-104 was prepared and stored at 4 °C in the dark and used within 5 days. Trametinib was dissolved in DMSO and resuspended in a solution of 0.5% Tween-80, 0.6% 2-hydroxypropyl (catalog no. H107-5G, Sigma-Aldrich) and 0.9% saline. Animals were treated with 0.25 mg kg−1 daily as described previously50. For the hemi-splenic studies, animals were enrolled in treatment 3 days after surgery and tissue was collected 14 days after surgery. For the combination treatment (DRP-104 and trametinib), tumors were collected after two cycles (two cycles of 5 days on, 2 days off) of treatment unless indicated otherwise. For the survival studies, either treatment modality continued until the animal reached the end point criteria. The end point criteria included but were not limited to abdominal hemorrhagic ascites, severe lethargy, cachexia, weight loss, poor posture, extreme weakness or unresponsive to external stimuli according to the IACUC approved protocol (no. IA16-00507).
In vivo metabolomics
For the in vivo metabolomics studies, HY19636 cells were orthotopically transplanted into B6J mice. Mice were treated with DRP-104 (3 mg kg−1) for either one or two cycles. Tumors were collected 30 min after the last DRP-104 dose; each tumor was snap-frozen in liquid nitrogen at the same time after euthanasia. Samples were subsequently processed at the Metabolomics Core Research Laboratory at NYU Langone Health. Briefly, frozen tissues were powdered with a biopulverizer (Biospec) and a fraction of the powder (approximately 10 mg) was transferred to a screw top vial for further mechanical homogenization with zircon beads using a BeadBlaster D2400 (Benchmark Scientific) at a fixed ratio of 20 mg ml−1 powder to extraction solvent comprising 80% LC–MS grade methanol containing isotopic internal standards (catalog no. MSK-A2-1.2, Cambridge Isotope Laboratories). Samples were homogenized for ten cycles at 30 ms and the resulting lysate was centrifuged at 21,000g for 3 min. Then, a fixed 450-µl volume of the metabolite supernatant was transferred to a new vial for speed vacuum concentration. The dried metabolite fraction was resolubilized in a fixed volume of 50 µl LC–MS grade water representing 1 mg of the original on-column tissue.
In vivo PDX efficacy studies
PDX samples from individuals with PDAC were used to test the in vivo efficacy of DRP-104. Materials were obtained under the participant’s consent and approved IRB (no. S17-00651) protocol at NYU Langone Health. NSG mice (female, 8–10 weeks old) were purchased from The Jackson Laboratory. All mice were bred and maintained at the animal facility of the NYU Grossman School of Medicine. All PDX samples were developed by direct engraftment of pancreatic cancer tissue fragments from patients undergoing surgical resection or biopsy into NSG mice, expanded and viably frozen. Except for sample NYU326, all PDX samples were therapy naive. Tumor fragments were implanted subcutaneously into NSG mice and expanded by passaging in the mouse without exposing samples to in vitro conditions. PDX was passaged in NSG mice to develop treatment cohorts. Mice were randomized and enrolled into each arm (vehicle versus DRP-104) once the tumor reached 2–4 mm diameter. Mice were treated with vehicle or 3 mg kg−1 DRP-104. Tumor length and width were measured every 5 days using a digital caliper. Tumor volumes were calculated using the formula (L × W2)/2. Animals were humanely euthanized when tumors reached more than 1,000 mm3 or at the end of four cycles of treatment.
PDO proliferation assays
PDO samples from individuals with PDAC were used to test the in vitro efficacy of DON. Materials were obtained under the participant’s consent and approved IRB (no. S17-00651) protocol at NYU Langone Health. All PDOs samples were developed by direct engraftment of pancreatic cancer tissue fragments from patients undergoing surgical resection or biopsy as domes in growth factor-reduced Matrigel, expanded and viably frozen at a low passage. PDOs were grown in PancreaCult Organoid Media (STEMCELL Technologies). PDOs were plated into 20 µl of growth factor-reduced Matrigel and supplemented with organoid media. PDOs were treated with DON at various doses 24 h after plating. PDO growth was assessed every 24 h using Cytation C10 for 7–10 days, depending on the growth kinetics of the PDO. Cell growth calculation was made based on the total optical density of each well.
Histology and IHC
Tumors were fixed in 10% formalin overnight and embedded in paraffin. Paraffin sections were deparaffinized and antigens were unmasked with citrate (pH 6) and heat. Slides were incubated in 3% hydrogen peroxide and 50% methanol for 30 min and blocked in 5% goat serum and 1% BSA in Tris-buffered saline with 0.05% Tween 20 (TBST) for 30 min at room temperature. Primary antibodies were diluted in blocking buffer and applied overnight at 4 °C, then developed using the VECTASTAIN Elite ABC-HRP kit (catalog no. PK-6100, Vector Laboratories) and DAB Substrate Kit (catalog no. SK-4100, Vector Laboratories). Slides were counterstained with hematoxylin (catalog no. H-3401, Vector Laboratories). Sections were dehydrated and mounted in Fisher Chemical Permount Mounting Medium (catalog no. SP15-100, Thermo Fisher Scientific) and allowed to dry overnight before slide imagining. The primary antibody was diluted in blocking buffer and added to sections at 4 °C overnight. Quantification of IHC was performed using the Aperio ImageScope software (Leica Biosystems); non-tumor and necrotic areas were not included in the analysis as described previously51. Briefly, we measured the percentage (%) of positivity resulting from the positive signal divided by the total area analyzed (Positive/AreaTotal).
To evaluate the role of CAF, 5-μm sections of paraffin-embedded tissue were stained with Akoya Biosciences Opal multiplex automation kit reagents unless stated otherwise. Automated staining was performed on a Leica Bond RX autostainer. The protocol was performed according to the manufacturer’s instructions with the following antibodies: CK19 (1:500 dilution, catalog no. MABT913, Merck Millipore), aSMA (1:4,000 dilution, catalog no. ab5694, Abcam), podoplanin monoclonal antibody (1:4,000 dilution, catalog no. 145381-82, eBioscience), PDGF R alpha (1:100 dilution, catalog no. AF1062, R&D Systems). Briefly, all slides underwent sequential epitope retrieval with the Leica Biosystems epitope retrieval 1 or 2 solution (citrate-based, pH 6.0, catalog no. AR9961; EDTA-based, pH9, catalog no. AR9640), primary and secondary antibody incubation and tyramide signal amplification with Opal fluorophores (catalog no. FP1487001KT, Akoya Biosciences). Primary and secondary antibodies were removed during the epitope retrieval steps while fluorophores were covalently attached to the epitope. Slides were mounted with ProLong Gold Antifade Mountant (catalog no. P36935, Thermo Fisher Scientific). Semiautomated image acquisition was performed on a Vectra Polaris multispectral imaging system. The inForm v.2.6 software from Akoya Biosciences was used for spectral unmixing and image analysis. To quantify the CAF subtypes, HALO (v.3.6, Indica Labs) was used. Briefly, a training algorithm was first used to identify nuclei (4,6-diamidino-2-phenylindole, nuclei segmentation AI module) on different samples during the training process. Then, the Highflex FL module algorithm (v.4.2.5, Indica Labs) was used to classify cells as CK19+, podoplanin+, aSMA+ and PDGFR+ using the phenotyper segmentation module. Each classification was reviewed for different samples during the training iterations. CAF subtypes were identified as pan-CAF (CK19−, podoplanin+); CK19−, podoplanin+, aSMA+, PDGFR− and CK19−, podoplanin−, aSMA+, PDGFR+. The thresholds for the classification was based only on high-expressing cells. Hematoxylin and eosin, Masson trichrome staining, slide scanning and Opal multiplexing were performed by the Experimental Pathology Research Laboratory at the NYU Grossman School of Medicine.
Western blot and antibodies
Whole-cell protein lysates were collected using radioimmunoprecipitation assay buffer (catalog no. 20-188, Sigma-Aldrich) supplemented with protease (Roche) and phosphatase (Roche) inhibitor cocktails. Protein lysates were separated on 4–20% gradient gels (catalog no. 4561096, Bio-Rad Laboratories) and transferred to polyvinylidene fluoride membranes (catalog no. IPVH00010, Merck Millipore) using transfer buffer (Tris-glycine) and 10% methanol. Membranes were blocked with 3% BSA (catalog no. A2058, Sigma-Aldrich) for at least 1 h at room temperature. Primary antibodies were incubated overnight at 4 °C at the following dilutions: ERK2 (1:1,000, catalog no. sc-1647, Santa Cruz Biotechnology); phospho-p44/42 MAPK (ERK1/2) (1:1,000, catalog no. 4376, Cell Signaling Technology); DUSP6 (1:1,000, catalog no. A2D4, Abcam); anti-O-linked N-acetylglucosamine (1:1,000, catalog no. ab2739, Abcam); cleaved Caspase-3 (1:1,000, catalog no. 9664, Cell Signaling Technology); ATF4 (1:1,000, catalog no. 11815, Cell Signaling Technology); phospho-eIF2α (Ser51) (1:1,000, catalog no. 3398, Cell Signaling Technology); Ki67 (1:500, catalog no. 16667, Abcam); GLUL (1:1,000, catalog no. 228590, Abcam); CK19 (1:100, catalog no. MABT913, Merck Millipore); anti-mouse IgG, horseradish peroxidase (HRP)-linked (1:5,000, catalog no. 7076 S, Cell Signaling Technology); anti-rabbit IgG, HRP-linked (1:5,000, catalog no. 7074 S, Cell Signaling Technology). For LI-COR imaging, IRDye 800CW goat anti-rabbit IgG (1:10,000, catalog no. 926-32211, LI-COR Biosciences) and IRDye 680RD goat anti-mouse IgG (1:10,000, catalog no. 926-68070, LI-COR Biosciences) secondary antibodies were used. Membranes were washed at least three times (15 min each) with TBST and incubated with the appropriate HRP-conjugated secondary antibody (1:5,000, catalog no. 7045S or 7076S, Cell Signaling Technology) for 1 h at room temperature; the enhanced chemiluminescence detection system was added (catalog no. 1705061, Bio-Rad Laboratories). Images were obtained with the ChemiDoc (Bio-Rad Laboratories) or Odyssey CLx (LI-COR Biosciences) imager. Each immunoblot was repeated at least twice.
Phospho-RTK array kit
The phosphorylation status of RTK was obtained using the Profiler Mouse Phospho-RTK Array Kit (catalog no. ARY014, R&D Systems) and Proteome Profiler Human Phospho-RTK Array Kit (catalog no. ARY001B, R&D Systems). Assays were performed according to the manufacturer’s instructions for each individual batch.
Bulk RNA-seq
Total RNA from cell cultures and tumors was obtained using TRIzol (catalog no. 15596026, Thermo Fisher Scientific) and PureLink RNA Mini Kit (catalog no. 12183025) according to the manufacturer’s instructions. Bulk RNA-seq libraries, raw data collection (FASTQ), alignment (HISAT2), mapping and differential expression analysis (DESeq2) were performed according to the manufacturer’s instructions by Novogene52. Logarithmic transformation (log2) of fragments per kilobase of transcript per million fragments mapped was used as input for GSEA53.
Statistics and reproducibility
All data were analyzed using Prism 9.0 (GraphPad Software). Results are expressed as the mean and s.e.m. unless otherwise indicated in the figure legend. The experiments were not randomized, and the investigators were not blinded to allocation during the experiments and outcome assessment. No statistical method was used to predetermine sample size. No data were excluded from the analyses. For the in vivo studies, before treatment initiation, mice bearing tumors were randomized. Tumor size was not measured at the time of randomization. For the tumor growth studies, the data shown represent independent experiments with biological replicates. For the in vitro studies, including the growth curves, Seahorse analysis and western blots were performed at least twice; the representative data of one experiment was presented. For the LC–MS and GC–MS, experiments were repeated twice with at least three biological replicates. IHC images represent a randomly selected image of a single biological sample. For all experiments, data distribution was assumed to be normal, but this was not formally tested. A Student’s two-tailed unpaired t-test was used for the experiments with two groups unless otherwise indicated. For multiple group comparisons, a one-way or two-way ANOVA was performed, followed by a Tukey or Holm-Šídák test unless otherwise indicated in the figure legend. The box plots extend from the 25th to 75th percentile. The whiskers represent the smallest to largest values. The line in the middle of the box represents the median. Data were considered significant if P < 0.05; exact values are shown in each figure legend or in the source data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.


留言 (0)