
記住我
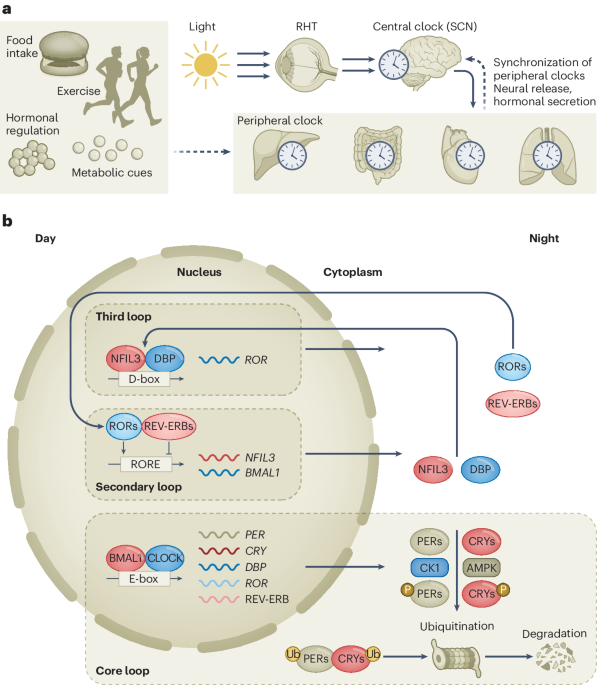
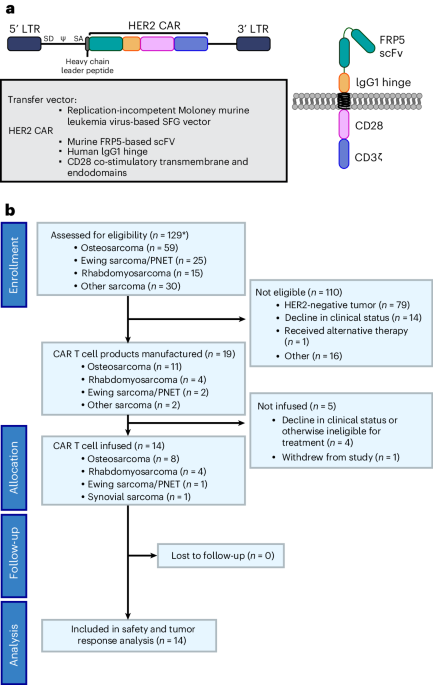

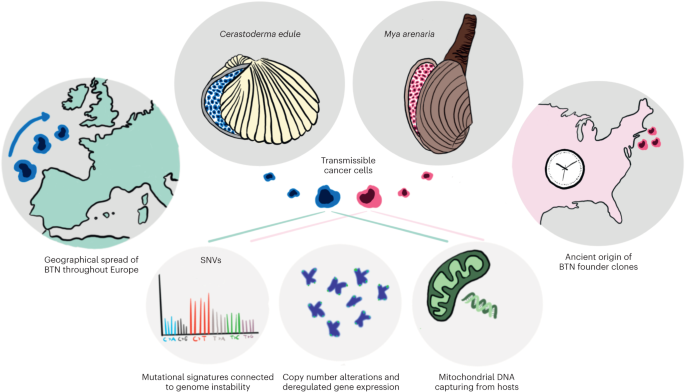
This study was approved by the Regional Committee for Medical and Health Research Ethics South-East Norway (2018/879, 2018/1246 and 2015/2357), the Institutional Review Board and the Data Protection Officer, Oslo University Hospital, the Swedish Ethical Review Authority, Stockholm (EPN 2017/2085-31/2) and the Ethical Committee in Central Denmark Region (1-45-70-88-21) and was performed in accordance with the Declaration of Helsinki. The Norwegian Food Safety Authority (application ID 17500) and Stockholms Djurförsöksetiska nämnd (17978-2018) approved all animal experiments.
Primary patient cells, healthy blood donor cells and cell linesBuffy coats (PBMCs) from healthy donors were provided by the blood bank of Oslo University Hospital, and PB or BM MNCs from patients with leukemia were isolated from cryopreserved, biobanked material (ethical approvals 2018/879 and 2018/1246). MNCs derived by density-gradient centrifugation (Axis-Shield) were stained to determine HLA-A2 expression by flow cytometry. To confirm the presence of FLT3D835Y, genomic DNA was extracted (QIAGEN DNeasy purification kit) from patient primary cells, and samples were sequenced using the TruSight Myeloid panel (Illumina). Information regarding the patients’ sex (reported in Supplementary Table 2) was not considered during the design of the study but was part of the overall clinical information obtained at the hospital. No sex- or gender-based analysis was performed because each patient is shown individually in the study.
Epstein–Barr virus-transformed lymphoblastoid cell lines (EBV-LCL) were generated from HLA-A2+ and HLA-A2− PBMCs as described previously27. All other cell lines were gifted or obtained from the American Type Culture Collection (ATCC) or the German Collection of Microorganisms and Cell Cultures (DSMZ) as indicated in the Nature Portfolio Reporting Summary. Authentication was performed by short tandem repeat DNA profiling by Labcorp DNA Identification Lab (formerly Genetica, https://celllineauthentication.com/). Cell line cultures were grown in humidified cell incubators containing 5% CO2 at 37 °C using media according to provider guidelines and were tested frequently for potential mycoplasma contamination.
Minigene designFor generation of T cell responses, a minigene was designed to encode predicted epitopes (https://services.healthtech.dtu.dk/service.php?NetMHC-4.0) containing the FLT3D835Y mutation, codon optimized and synthesized by GenScript. Subsequently, it was cloned into the pCIpA102 vector for in vitro mRNA transcription using the RiboMAX Large Scale RNA production system (Promega), as previously described69,70. The minigene encoded the FLT3 amino acid sequence KICDFGLARYIMSDSNYVVRGNVRLARLP (FLT3D835Y 9-mer and 10-mer are underlined and the mutation in position 1 is shown in bold).
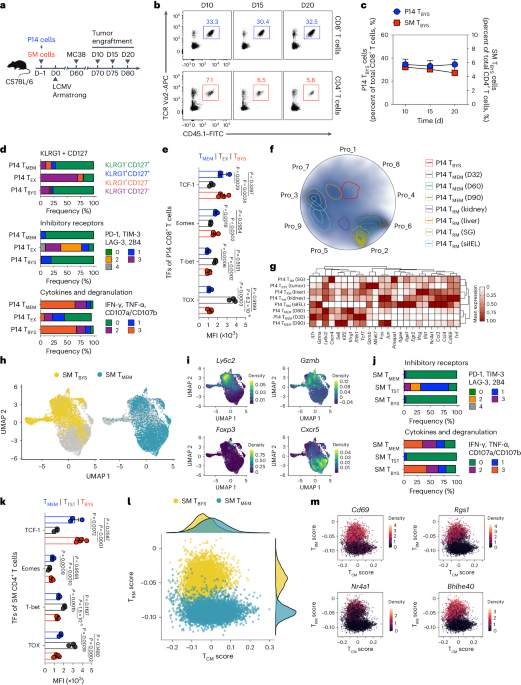
Induction of antigen-specific T cellsMonocytes from HLA-A2+ healthy donors were isolated on day −4 using CD14-reactive microbeads and the autoMACS Pro Separator (Miltenyi Biotec). The CD14− PBMC fraction was cryopreserved for later use. The monocytes were then cultured for 3 d in CellGro GMP DC medium (CellGenix) with 1% (vol/vol) human serum (HS; Trina biotech), 1% (vol/vol) penicillin–streptomycin (P/S; Sigma-Aldrich), 50 IU ml−1 IL-4 (PeproTech) and 800 IU ml−1 GM-CSF (Genzyme). Subsequently, moDCs were matured for 14–16 h by adding lipopolysaccharide (Sigma-Aldrich) and IFN-γ (PeproTech) to final concentrations of 10 ng ml−1 and 100 IU ml−1, respectively. On day −1, naive CD8+ T cells were isolated from the autologous CD14− cryopreserved PBMCs by use of the autoMACS Pro Separator and a CD8+ T cell-isolation kit, into which CD45RO- and CD57-reactive beads (Miltenyi Biotec) were added. On day 0, moDCs were collected, electroporated with mRNA and co-cultured with naive T cells in DC–T cell medium with 30 ng ml−1 IL-21 (PeproTech) at a DC:T cell ratio of 1:4. After 10 d, co-cultures were screened for the presence of FLT3D/Y pMHC multimer-reactive CD8+ T cells. pMHC multimers labeled with PE and APC were prepared in house as described previously71,72. Viable CD8+pMHC+ T cells (double positive for PE- and APC-conjugated pMHC multimers) were sorted by flow cytometry.
Expansion of memory T cells reactive to FLT3D/Y in samples from patients with AML following HSCTFor in vitro expansion of potential memory T cells reactive to the FLT3D/Y-mutant peptide in patients with AML that had undergone HSCT, cryopreserved PB samples were thawed and resuspended in Iscove’s Modified Dulbecco’s Medium (IMDM) with 20% (vol/vol) FCS (Trina biotech) and 0.1 mg ml−1 DNase. Viable cells were resuspended at a concentration of 1 M ml−1 and pulsed with the FLT3D/Y peptide at 100 ng ml−1 for 2 h at 37 °C. Cells were washed and resuspended at 3.75 million cells per ml in IMDM with 5% HS, 1× P/S and 20 U ml−1 IL-2 before culturing for 5 d. Identification of T cells reactive to the peptide was performed by staining with pMHC multimers as described above.
Sorting and cloning of pMHC multimer+CD8+ T cellsPBMCs from three healthy donors were mixed at an equal ratio (1:1:1) and irradiated with 35 Gy, washed and resuspended in X-VIVO 20 medium (Lonza, BioNordika) with 5% HS and 1% P/S (T cell medium). A total of 0.2 × 106 irradiated cells (feeders) were placed into tissue culture-treated 96-well plates and were supplemented with 100 μl T cell medium containing 2 μg ml−1 phytohemagglutinin (Remel Thermo Scientific), 80 ng ml−1 IL-2 (R&D Systems) and 4 ng ml−1 IL-15 (PeproTech). FLT3D/Y co-cultures were then collected and stained with LIVE/DEAD Fixable Near-IR, anti-CD3 antibody, anti-CD8a antibody and PE- and APC-conjugated pMHC multimers, and, using the FACSAria II (BD Biosciences) cell sorter, CD8+, pMHC-double-positive multimer populations were sorted as single cells into 96-well plates containing feeders. After 7 d, cultures were supplied with fresh T cell medium containing 1,750 U ml−1 IL-2 and 4 ng ml−1 IL-15, and expanding clones were identified by microscopy. On day 14, growing clones were restimulated with feeder cells prepared as described above and stained with FLT3D/Y pMHC multimers. To assess functionality after expansion, clones were stimulated with K562 cells pulsed with FLT3D/Y or FLT3WT peptides and assessed for CD137 upregulation.
TCR sequencingThe sequences for paired TCR-α and TCR-β chains from two clones and 55 single cells reactive to FLT3D/Y pMHC multimers were amplified as previously described but modified and adapted for the targeted amplification of transcripts encoding TCR-α and TCR-β (refs. 27,38,55). The MiXCR script was used to analyze sequencing data, and an in-house Python script TCR primer was used to reconstruct full-length TCR chains as described previously55,73. The output was manually verified using IMGT/V-QUEST74. For identified TCRs, codon-optimized sequences for TCR-α and TCR-β variable fragments were synthesized and cloned by GenScript.
Gene transfer to human PBMCs and cell linesHLA-A2+ healthy donor-derived and patient-derived PBMCs were transduced to express FLT3D/Y- and NY-ESO-1 (1G4)-specific TCRs, as detailed in ref. 27. Briefly, 2 × 106 PBMCs per ml in CellGro GMP DC medium with 5% (vol/vol) HS, IL-7 and IL-15 (5 ng ml−1 each, PeproTech) were added to antibody-coated plates (anti-CD3 clone OKT3, eBioscience and anti-CD28 clone CD28.6, eBioscience) and incubated at 37 °C with 5% CO2 for 72 h. Retroviral supernatants were generated as described previously27. PBMCs were collected, resuspended in CellGro GMP DC medium with 5% HS, IL-7 and IL-15, mixed with retroviral supernatant, placed in non-tissue culture-treated six-well plates precoated with RetroNectin (20 μg ml−1, Takara) and spinoculated at 900g for 60 min twice on consecutive days. Transduction efficiency was determined after 3 d by staining with anti-mouse TCR-β chain antibody and/or the pMHC multimer followed by flow cytometry. Before functional experiments, cells were cultured for 48–72 h in CellGro GMP DC medium containing low concentrations of cytokines (0.5 ng ml−1 IL-7 and IL-15). Alternatively, cells were frozen for later experiments.
BV173, ML-2, RS4;11 and NALM-6 cell lines were transduced as described above using retroviral supernatant containing the FLT3D/Y minigene. For in vivo experiments, the BV173 cell line was stably transduced to express the FLT3D/Y minigene, firefly luciferase and GFP (hereafter, BV173D835Y). Complementary DNA encoding FLT3D/Y and HLA-A2 was cloned into the pCIpA102 vector for production of mRNA, as previously described70,71
Immunoprecipitation-targeted mass spectrometry analysis of FLT3 peptides presented on HLAMonoallelic B721.221 cells expressing HLA-A2 were transduced to express the mutant FLT3 amino acid sequence VLVTHGKVVKICDFGLARYIMSDSNYVVRGNARLPVK. One hundred million cells from the B721.221 line and 400 million cells from patient samples (patients 1 and 3) were lysed in PBS containing 1% lauryl maltoside, 0.5 mM EDTA, 1 mM PMSF and Sigma protease inhibitors (1:200) for 1 h at 4 °C (1 ml lysis buffer per 100 million cells). The clarified cell lysates were then added to 200 µl of AminoLink Plus bead slurry (Thermo Fisher Scientific) coated with pan-HLA class I-specific antibody (W6/32, BioXCell) to enrich for HLA peptides27. The HLA-bound peptides were then sequentially eluted three times, each with 1 ml of 1% TFA. Peptide elutions were pooled and desalted using the Discovery DSC-C18 SPE column. The peptides were vacuum concentrated and dissolved in 25 μl of 3% acetonitrile containing 0.1% TFA, following spike-in with 200 pg of heavy isotope-labeled peptide (YI(13C6,15N)MSDSNYV). The peptide solution (5 μl) was analyzed using an EASY-nLC 1000 system (Thermo Fisher Scientific) connected to a Q Exactive HF mass spectrometer (Thermo Electron) equipped with a nano-electrospray ion source. For liquid chromatography separation, an EASY-Spray ES902 column (C18, 2-µm beads, 100 Å, 75-μm inner diameter) capillary with a bed length of 25 cm was used. A flow rate of 300 nl min−1 was employed with a solvent gradient of 7–35% B in 55 min to 90% B in 3 min. Solvent A was 0.1% formic acid, and solvent B was 0.1% formic acid–90% acetonitrile. The mass spectrometer was operated in parallel reaction-monitoring mode to specifically target the presence of endogenous FLT3 mutant (m/z = 1,091.47381+) and spiked-in isotope-labeled peptide (m/z = 1,098.49281+), eluting within a retention time window of 27–31 min, as determined using a synthetic analog. The MS/MS spectra using higher-energy collision-induced dissociation were acquired with a resolution of R = 15,000 after accumulation to a target of 1 × 105. The normalized collision energy was set to NCE 27, and the isolation window was m/z = 2.0. The maximum allowed ion accumulation for the MS/MS spectrum was 120 ms. Raw data were analyzed using Xcalibur software and Skyline (MacCoss Lab Software).
pMHC-stability assayHLA-A2 molecules were prepared in house, as previously described71,75,76. The pMHC-stability assay was performed as previously described38 with minor modifications. UV-mediated peptide-exchange reactions were performed for 1 h, followed by incubation of the resulting product at 4 °C. The next day, streptavidin-coated beads were washed twice with PBS–1% Tween. The peptide–HLA monomers were coupled with the washed beads for 10 min at room temperature. After coupling, the beads were washed twice with PBS–1% Tween and resuspended in 200 μl. An aliquot of 20 μl beads was set aside for the 0-h time point, while the remaining beads were incubated at 37 °C. Twenty microliters of beads were collected at 3, 6, 12 and 24 h of incubation. After collecting at each time point, the beads were stained with 30 μl of anti-HLA-A2–PE antibody (343305, BioLegend, 1:100 dilution) for 10 min at room temperature. Samples from all time points were analyzed immediately after staining on a BD LSR II Flow Cytometer.
Antibodies, dyes and flow cytometryFor surface antibody staining of human PB and BM cells, antibodies were added to cells for 15–20 min at 4 °C, followed by washing steps. For intracellular staining, cells were suspended in Cytofix/Cytoperm (BD Biosciences) solution for 20 min, washed with Perm/Wash buffer (BD Biosciences) and then stained with antibodies. For mouse PB, BM and spleen, cells were processed into a single-cell suspension as previously described77 and Fc receptor blocked (human, Miltenyi Biotec; mouse, produced by mouse hybridoma cell line clone 2.4 G2, ATCC, HB-197) for 10 min at 4 °C before staining with antibodies for 15–20 min at 4 °C. All fluorescently conjugated antibodies are described in Supplementary Table 10 and the Nature Portfolio Reporting Summary. In PDX mice, the percentage of myeloid cells was determined as a fraction of combined mouse and human leukocytes (mCD45+hCD45+), subtracting hCD3+ events accounting for infused T cells. Flow cytometry analysis was performed on the BD LSR II flow cytometer or the BD LSRFortessa machine (both BD Biosciences), while cell sorting was performed on the FACSAria Fusion cell sorter (BD Biosciences). Data were analyzed using FlowJo (TreeStar) or FACSDiva (BD Biosciences) software. To visually display flow cytometry data, we used an unsupervised nonlinear dimensionality-reduction algorithm such as t-SNE by using FlowJo (TreeStar) software.
T cell-activation assaysT cells transduced to express TCR were co-cultured with cell lines or primary patient tumor cells at an E:T cell ratio of 1:2 (100,000:200,000 cells per well), and reactivity was investigated by measuring CD137 upregulation or IFN-γ release. When indicated, target cells were pulsed with FLT3D/Y or FLT3WT peptide (purities >90%) or 161 single-amino acid-substituted variants of the FLT3D/Y peptide (purity >70%) (GenScript Biotech) for 1–2 h or electroporated with mRNA encoding either FLT3WT or the FLT3D/Y minigene. Cells were placed in round- or flat-bottom 96-well plates and, after 18–20 h of co-incubation, were centrifuged at 700g for 2 min. Supernatants were collected for measurement of IFN-γ levels by ELISA, while cells were washed and stained with anti-CD137 antibody. In some experiments, transduced T cells were labeled with 0.75 μM CTV to distinguish them from target cells. Reagents for the IFN-γ ELISA were acquired from BD Pharmingen or R&D Systems: mouse anti-human IFN-γ capture antibody (NIB42), Biotin Mouse Anti-Human IFN-γ-detection antibody (4S.B3), streptavidin–HRP, stabilized tetramethylbenzidine and hydrogen peroxide as substrate solutions, sulfuric acid as the stop solution and recombinant human IFN-γ protein as the standard. The assay was performed according to the manufacturer’s instructions.
Flow cytometry-based cytotoxicity assay using cell lines as targetsTransduced cell lines stably expressing the FLT3D/Y minigene were co-cultured with CTV-labeled T cells transduced to express TCR at an E:T cell ratio of 1:2 (75,000:150,000 cells per well) for 48 h in round-bottom 96-well plates in triplicates. Following co-culture, cells were collected, washed and stained with human anti-CD3, anti-CD8 and anti-CD4 antibodies and LIVE/DEAD NIR for 15–20 min. Cells were then washed and resuspended in FACS buffer containing 10,000 CountBright Absolute Counting Beads (Thermo Fisher). An equal number of bead events (3,500) were recorded from every well. Normalized data were reported as percentage of the mean of the number of viable tumor cells acquired from three parallel wells co-cultured with TCR1G4 or TCRFLT3D/Y cells from each donor.
Flow cytometry-based assays for T cell activation and cytotoxicity using primary human samplesPB or BM samples from patients were thawed and resuspended in IMDM with 20% (vol/vol) FCS (Trina biotech) and 0.1 mg ml−1 DNase. Cells were centrifuged at 200g for 15 min at room temperature and transferred to round-bottom 96-well plates for assays measuring CD137 upregulation on T cells transduced to express TCR or cytotoxicity on target cells. Normal CD19+ B cells were isolated from healthy donor buffy coat MNCs using CD19-reactive microbeads and the autoMACS Pro Separator (Miltenyi Biotec) and transferred to round-bottom 96-well plates for assays measuring cytotoxicity on target cells. Individualized antibody panels and gating strategies to identify malignant blasts and normal leukocyte populations were designed after reviewing diagnostic phenotyping available in the hospital records. Allogeneic or, for patients 1–3, also autologous patient-derived T cells transduced to express TCRs, were used in the experiments. Cells transduced to express TCR were prelabeled with CTV dye to distinguish them from target cells. For cytotoxicity assays, 75,000 T cells per well were co-incubated with 150,000 target cells in three parallel wells per condition for 72 h and then stained with individualized antibody panels for flow cytometry. CountBright Absolute Counting Beads were used as described above. Examples of the gating strategy used to identify live tumor cells in patients are shown in Extended Data Fig. 6a. For patients 1–6 and 8, myeloid cells were identified as CD3−CD19−CD20−, normal T cells as CD3+ and normal B cells as CD19+CD20+. Cells from patient 7 were obtained from Jackson Laboratory (stock ID J000106565), and the patient-specific phenotypic markers CD33 and CD19 were used to identify leukemia cells based on the characterization profile from the provider.
TCRFLT3D/Y cell activity in the xenograft leukemia cell line modelThis study was approved by the Norwegian Food Safety Authority (application ID 17500) and performed in compliance with institutional guidelines and the 2010/63/EU directive. Mice were observed for clinical signs of tumor spreading and were killed if they developed >20% weight loss, hunched posture, ruffled fur, limb paralysis or enlarged spleens. Maximum tumor burden was not exceeded. Experiments were terminated 2 months after T cell injection to avoid graft-versus-host disease, and surviving mice were killed humanely by cervical dislocation. Six mice were housed per cage in Eurostandard Type III cages (macrolone) with a light cycle from 7 a.m. to 7 p.m. at 22 ± 1 °C with 62 ± 5% humidity. Female (8–10-week-old) NSG (Jackson Laboratory) mice, bred in house, were sublethally irradiated (2.5 Gy, MultiRad225 X-ray, RPS Services) on day −15, and 4 × 106 cells of the human B-ALL cell line BV173D835Y were injected on day −14 through the tail vein. After leukemia was confirmed by BLI on day −1, mice were treated with 107 T cells transduced to express TCR1G4 or TCRFLT3D/Y. A group of control mice did not receive T cell injections. All mice were injected intraperitoneally (i.p.) daily with 2,500 IU IL-2 (R&D Systems). BLI imaging (by the IVIS Spectrum in vivo imaging system; analysis with Living Image software version 4.5.2, PerkinElmer) and blood analysis by flow cytometry were performed continuously. BM was collected at the endpoint and processed for flow cytometry to analyze the presence of T cells and tumor cells.
Activity of TCRFLT3D/Y cells in four primary AML PDX modelsExperiments were approved by Stockholms Djurförsöksetiska nämnd (17978-2018). The maximal tumor burden permitted was defined by the impact on the animal’s health. Mice engrafted with leukemic cells were continuously monitored according to Karolinska Institutet’s health assessment, and no animal exceeded the humane endpoint. The housing conditions were 21 °C and 45–50% humidity. Two to five mice were housed per cage in IVC-Mouse GM500 cages with a light cycle from 4 a.m. to 4 p.m. (patient 7) or from 6 a.m. to 6 p.m. (patient 1). BM was collected from femur, tibia and crista from both hind legs at terminal analysis. Mainly female mice were used in this study, but, when both sexes were used, they were equally distributed within the treatment groups. No sex-based analysis was thus performed.
Model 1Female NOD.Cg-PrkdcscidIl2rgtm1Wjl Tg(CMV IL3,CSF2,KITLG)1Eav/MloySzJ (NSG-SGM3) mice stably engrafted with FLT3D835Y-expressing HLA-A2+ AML cells from patient 7 (Supplementary Table 2 and 3) at 5 weeks of age were obtained from Jackson Laboratory (stock ID J000106565). Upon arrival (5.5 weeks after transplantation), PB myeloid engraftment was confirmed by flow cytometry, and mice were allocated to treatment groups (TCRFLT3D/Y or TCR1G4 cells), resulting in a similar mean human myeloid engraftment between the groups before infusion with T cells. Cryopreserved T cells transduced to express TCR were thawed and cultured in X-VIVO 20 medium (Lonza) with 5% HS, 1% penicillin–streptavidin and 5 ng ml−1 IL-7 and IL-15 for 3–4 d before treatment. T cells containing 5 × 106 CD8+mTCR-β+ T cells were injected through the lateral tail vein, and all mice received daily i.p. injections of 2,500 IU human IL-2 (R&D Systems). At day 8 after T cell infusion, half of the mice received a second dose of 5 × 106 CD4-depleted (Miltenyi Biotec) TCRFLT3D/Y or TCR1G4 T cells. As no differences were observed between the mice receiving one or two doses of T cells, the data from these mice were pooled. The effect of the T cells was monitored in serially collected PB and at termination 15 d after T cell treatment in the BM and spleen through detailed flow cytometry analysis.
Model 2BM CD34+ cells from patient 1 (FLT3D/Y and HLA-A2+; Supplementary Tables 2 and 3) were obtained by CD34 magnetic bead enrichment (Miltenyi Biotec) according to the manufacturer’s instructions and as previously described78. A total of 3 × 105 CD34+ cells per mouse were intrafemorally injected into sublethally irradiated (3.3 Gy, X-ray source) female and male NSG-SGM3 mice (Jackson Laboratory, stock 013062) 9 weeks of age. Upon confirmation of stable PB myeloid engraftment 7 weeks after transplantation, mice were allocated to treatment groups (TCRFLT3D/Y or TCR1G4 cells) based on their engraftment levels as described for patient 7. T cells containing 5 × 106 CD8+mTCR-β+ T cells were infused into each mouse by lateral tail vain injections, and all mice received daily i.p. injections of 2,500 IU human IL-2 for 2 weeks, followed by less frequent injections. Mice were monitored using serially collected PB and at termination 34 d after T cell treatment with the BM and spleen.
Model 3To mimic an MRD setting, secondary intrafemoral transplantation of BM cells from NSG mice (Jackson Laboratory, stock 005557) engrafted with material from patient 1 was performed into sublethally irradiated (2.5 Gy, X-ray source) female NSG mice 12–13 weeks of age. Following confirmation of low but stable leukemic engraftment 20 weeks after transplantation, mice were treated with TCRFLT3D/Y or TCR1G4 CD4-depleted (Miltenyi Biotec) T cells (10 × 106 T cells, containing 90% CD8+ and 10% CD4+ T cells). All mice were injected i.p. daily with 2,500 IU human IL-2. The effect of the T cells was evaluated in the BM 11 d after T cell treatment.
Model 4Following secondary transplantation of BM from mice engrafted with patient 1 cells, BM from three NOD.Cg-PrkdcscidIl2rgtm1Sug Tg(CMV-IL2)4-2Jic/JicTac (NOG-hIL2, Taconic) mice was isolated and cultured for 48 h in CellGro GMP DC medium with 5% HS, IL-7 and IL-15 either with no T cells or with TCR1G4 or TCRFLT3D/Y cells (E:T cell ratio, 1:2). After 48 h, the contents of each well were collected and intrafemorally injected into sublethally irradiated (2.25–2.5 Gy) female NSG mice 8–13 weeks of age. Engraftment levels were monitored in serially collected PB samples by flow cytometry. Data were pooled from two individual experiments. Differences in engraftment dynamics between mice engrafted with AML cells precultured with TCRFLT3D/Y or TCR1G4 cells or without T cells was assessed by multilevel linear regression using the R package ‘lmerTest’. Sampling time points and groups are treated as an interaction term with random effects for individual mice. Because the engraftment at the first time point was 0 for all mice except one, we fixed the intercept as 0. Estimated marginal means of models were obtained and compared between three groups using the R package ‘emmeans’. Multiple tests were corrected using the Benjamini–Hochberg method. Only mice that could be followed until the end of the experiment were included in the analysis.
Whole-exome sequencing of cells from patient 1 and PDX miceDNA was isolated from AML cells purified with the FACSAria Fusion cell sorter (BD Biosciences) from patient 1 using the Maxwell RSC Cultured Cells DNA Kit (Promega), including primary BM AML blasts (CD3−CD19−) and T cells (CD3+CD8+CD19−CD33− and CD3+CD4+CD19-CD33-) and BM mCD45−hCD45+CD3−CD19− cells from one untreated and one TCR1G4-treated PDX mouse from model 2. Whole-exome sequencing libraries were prepared with the Lotus DNA Library Prep Kit (IDT). Exon regions were captured using xGen Exome Hyb Panel v2 and the xGen Hybridization and Wash Kit (IDT) on 4 July 2022 and 5 July 2022. After mixing with 1% PhiX, prepared libraries were sequenced using the NextSeq high-output kit (300 cycles) with settings ‘Read1, 151; Index1, 8; Index2, 8; Read2, 151’. Reads were aligned to the human (GRCh37) genome reference using Burrows–Wheeler Aligner version 0.7.17 with default parameter settings. PCR duplicates were marked with biobambam version 2.0.87. Reads were subjected to indel realignment and base quality score recalibration using GATK3 (version 3.8) and recalculation of MD/NM tags using SAMtools version 1.9. Errors associated with enzymatic fragmentation during library preparation were removed using FADE version 0.5.5, resulting in an average depth of 364 (331–439) in the final bam files79.
Mutations calling was performed using GenomonFisher (https://github.com/Genomon-Project/GenomonFisher) with the following parameters: (1) mapping quality score ≥20, (2) base quality score ≥15, (3) number of total reads ≥8, (4) number of variant reads ≥5, (5) VAF ≥ 0.05, (6) VAF in paired T cells <0.1, (7) VAF in other non-paired normal controls <0.05 in all and average <0.01, (8) strand ratio in tumor ≠0 or 1, (9) VAF by base counts divided by VAF by read count ≥0.5 and ≤2, (10) P value by Fisher <0.1, (11) P value by EBFilter80 <0.001, (12) mutation on exons, (13) not on the repeat regions.
Mutations were annotated by ANNOVAR81. Copy number analysis was performed using CNACS82. The WT1H507P mutation was, together with the FLT3D835Y mutation, the only identified potential driver mutation in AML15,83.
Droplet digital PCRddPCR was performed to quantify clonal involvement. DNA from patient 1 with AML was isolated as explained above, and DNA from AML PDX mice was isolated through flow cytometry sorting of mCD45−mTer119−hCD45+CD3− cells (PDX patient 7) or hCD45+CD33+CD34−, hCD45+CD33+CD34+ and hCD45+CD19+ cells (PDX patient 1) and subjected to whole-genome DNA amplification using the REPLI-g Single Cell Kit (QIAGEN) according to the manufacturer’s instructions. Samples with <90 cells were excluded. Briefly, a 20-µl PCR reaction mixture containing 1× ddPCR supermix for probes (no dUTP) (Bio-Rad), 1× primer-probe assay (FLT3D835Y, dHsaMDV2010047; WT1H507P, dHsaMDS871718945; Bio-Rad) and 60 ng DNA was mixed with Droplet Generation Oil for Probes (Bio-Rad). Droplets were prepared according to the manufacturer’s instructions on a QX200 droplet generator (Bio-Rad) and subjected to PCR (Bio-Rad): 95 °C for 10 min, 40 cycles of 94 °C for 30 s and 55 °C for 60 s and a 10-min incubation at 98 °C. Plates were read on a QX200 droplet reader (Bio-Rad) and analyzed using QuantaSoft version 1.5.38.1118 software (Bio-Rad) to calculate VAFs and 95% confidential intervals. The numbers of FLT3D835Y cells in the AML PDX mice were determined by combining the information from the fraction of mutated cells identified by ddPCR, and the frequency of phenotypically defined subsets (hCD45+CD3− or hCD45+CD33+CD34− and hCD45+CD33+CD34+, respectively, for patients 7 and 1) in the BM as determined by flow cytometry, and the total number of MNCs in the BM was counted with Sysmex.
External AML-targeted sequencing data analysisMutation data (SNVs and indels) from AML patients reported by Papaemmanuil et al.15 were downloaded from https://www.cbioportal.org. VAF was estimated from reported alternative allele reads divided by sequencing depth for the position. Patients harboring a FLT3D835Y mutation were selected for in-depth analysis.
The order of FLT3 mutations in AML using VAF and the mutated cell fraction was determined from the publicly available mutation list identified by targeted DNA sequencing by Morita et al.45.
Statistical analysis and reproducibilityStatistical analysis was performed in GraphPad Prism versions 6–8 (GraphPad Software). Comparison of mean values between two experimental groups was conducted with unpaired, two-tailed Student’s test. The ordinary ANOVA test with adjustment for multiple comparisons with Tukey’s post hoc test was employed for comparisons of more than two experimental groups. To determine differences between in vivo treatment groups in the PDX mouse models, Kruskal–Wallis ANOVA with Dunn’s multiple-comparison test and two-tailed Mann–Whitney test were performed. P values < 0.05 were considered statistically significant. The investigators were not blinded to allocation during experiments or to outcome assessment. PDX models 1–3 were performed once each, and data for PDX model 4 were generated from two independent experiments with mice from all treatment groups represented in both experiments. No statistical method was used to predetermine sample size, and experiments were not randomized. Sample sizes were estimated based on preliminary experiments and are similar to those previously reported27,84. Due to the nature of the other in vitro experiments, blinding was not possible and is not generally performed in the field as the data acquisition is quantitative (flow cytometry or MS) rather than qualitative and therefore less influenced by observer bias. Data distribution was assumed to be normal, but this was not formally tested. Data distribution is shown as individual data points.
IllustrationsFig. 1a was generated by Science Shaped (https://scienceshaped.com/). All mouse illustrations were generated using Adobe Illustrator 2022 version 26.0.3.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)