
記住我

Memory deficits and spatial disorientation of the index patient were first noted at the age of 53 years. At the age of 57 years, he presented for the first time to our clinic with spatial and temporal disorientation, forgetfulness, reduced motivation, disinhibition, and socially inadequate behavior. The basic neurological examination was normal. 21/30 points were achieved in the MMSE. Further neuropsychological testing revealed prominent deficits in episodic memory as well as executive deficits concerning planning, conceptualization, abstract thinking, flexibility, and monitoring. He showed disinhibited behavior and anosodiaphoria. In the following 3 years, all deficits progressed further with addition of visuoconstructive deficits and apraxia as well as reduced drive and attentional deficits.
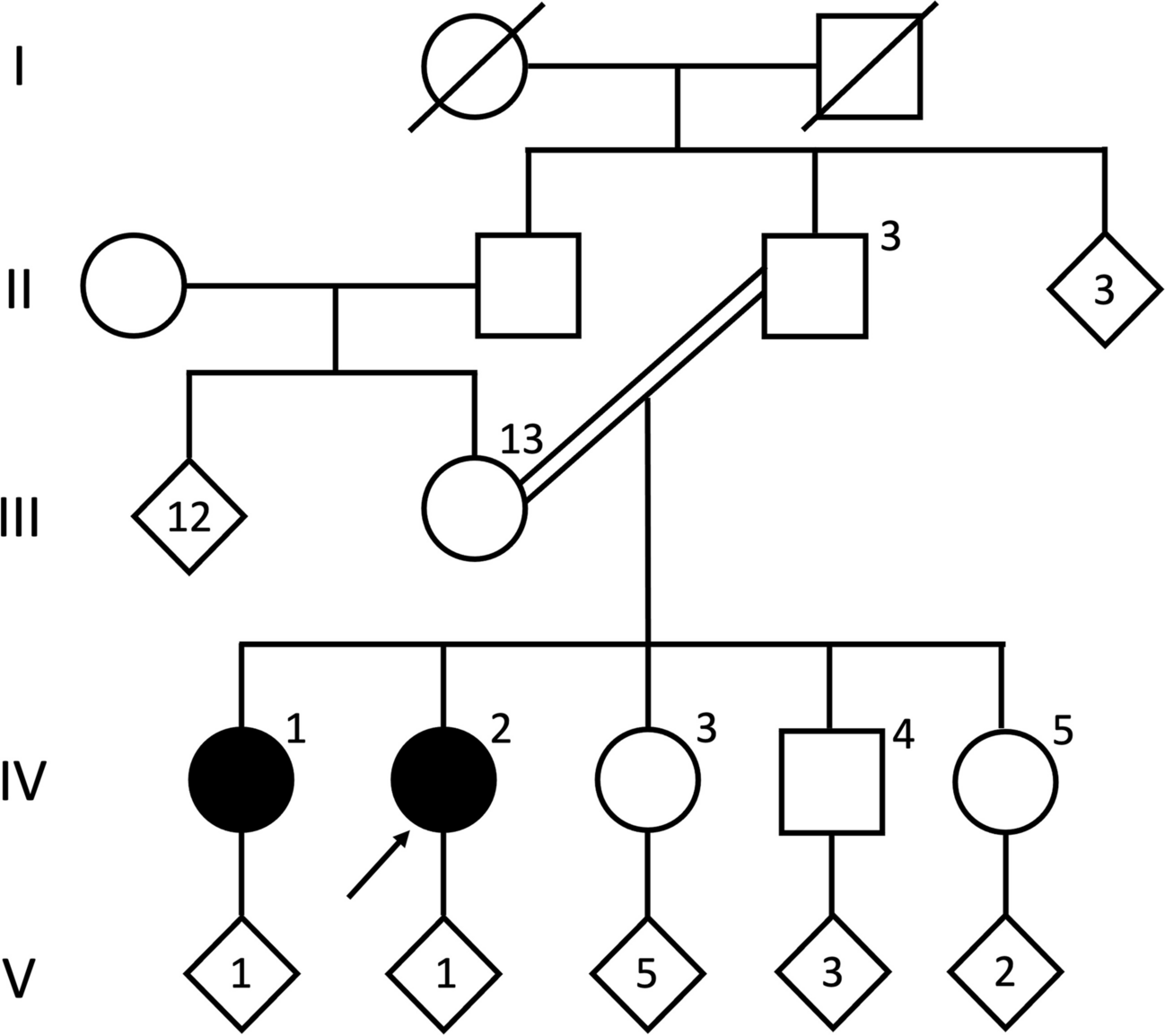
CT and MR imaging revealed symmetrical calcifications in the pallidum, thalamus, cerebellum, and also the hippocampus (Fig. 1A–E), suggestive of PFBC. Whole exome sequencing and targeted analysis of known PFBC genes followed by Sanger sequencing confirmation (Suppl. Figure 1) revealed a heterozygous variant in SLC20A2 (c.1523 + 1G > T), which is predicted to disrupt a splice donor site and lead to a translational frameshift. RT-PCR and Sanger sequencing of the PCR product was successfully used to detect wild-type but failed to identify mutant SLC20A2 mRNA from patient whole blood (data not shown). This observation could be explained by the generally low expression of SLC20A2 in blood cells ([8]; www.proteinatlas.org/ENSG00000168575-SLC20A2/single+cell+type) possibly together with nonsense-mediated RNA decay of the mutant mRNA. Taking together the symmetric brain calcifications and the fact that loss-of-function mutations in SLC20A2 have repeatedly been shown to cause PFBC, it is highly likely that the SLC20A2 variant is causative. Causality was further corroborated by segregation of the mutation with milder pallidal calcifications in two children of the patient, which were absent in a third child who was tested negative for the mutation (Fig. 1F–H). All three children were of full age but younger than 30 years. One of the two children carrying the SLC20A2 mutation (Fig. 1F) suffers from panic disorder and depression but has no movement disorder or cognitive deficits (29/30 points in the MMSE). The other SLC20A2 mutation carrier (Fig. 1G) is asymptomatic and achieved also 29/30 points in the MMSE. The third child, which was tested negative for the mutation (Fig. 1H), suffers from depression and 30/30 points were scored on the MMSE.
Fig. 1
Axial cerebral CT and susceptibility-weighted MR imaging of the index patient (A–E) shows extensive calcifications of bilateral basal ganglia (in pallidum and caudate nucleus) as well as in both cerebellar hemispheres (arrows) and bilateral hippocampus (arrowheads). Susceptibility-weighted MR imaging of the index patient’s children (F–H) shows symmetrical calcifications of the basal ganglia (arrows) in both child 1 and child 2. Child 3 shows no calcifications. Axial and coronal FDG-PET/CT (I, J) of the index patient demonstrates areas of slightly reduced glucose metabolism in parietal and mesiotemporal cortex. FBB-PET/CT (K, L) study in axial and coronal sections shows widespread β-amyloid deposits in the frontal and temporal cortex but also in the parietal cortex. Red/light red indicates high values; green/blue indicates low values
Secondary basal ganglia calcifications were excluded in the index patient. CSF analysis revealed normal values for total protein, CSF/serum albumin ratio, cell count, and lactate; there was no intrathecal IgG synthesis. In contrast, CSF tau, phospho-tau, and β-amyloid protein levels were compatible with Alzheimer’s disease. Remarkably, at the age of 57 years, i.e., 4 years after onset of symptom, CSF Tau protein and β-amyloid values had still been in the normal range (total tau protein 272 pg/ml (normal range < 445 pg/ml); β-amyloid 381 pg/ml (> 375 pg/ml)) or only marginally elevated (phospho-tau 68 pg/ml (< 61 pg/ml)). At 60 years of age, however, all markers were pathological (total tau protein 777 pg/ml (normal range < 400 pg/ml); phospho-tau 107 pg/ml (< 60 pg/ml); β-amyloid 1–42 359 pg/ml (> 600 pg/ml); β-amyloid 42/40 ratio 0.05 (> 0.07)). Consequent FDG-PET imaging showed a mildly reduced glucose metabolism in parietal and mesiotemporal cortical regions (Fig. 1I, J). Moreover, a FBB-PET study demonstrated widespread cortical β-amyloid deposits in cortical regions (Fig. 1K, L). Considering the CSF analysis and PET imaging results that were in agreement with Alzheimer’s disease pathology, re-analysis of the whole exome data was performed and revealed the genetic missense variant c.235G > A/p.A79T in the EOAD gene PSEN1 that was confirmed by Sanger sequencing (Suppl. Figure 1). The variant alters a highly conserved amino acid and is predicted to be damaging (CADD score = 33; predicted to be “probably damaging” by polyphen2). The index patient’s children were all tested negative for the mutation.
The rarity of this variant in reference databases (gnomAD allele frequency 4/251,404) and the fact that another missense mutation in the same codon has been observed before in three other families affected by EOAD [9] further corroborates its causality, and amino acid 79 of PSEN1 could represent a mutational hotspot. The APOE genotype was found to be ɛ3 (homozygous reference sequence) in this patient. The mother of the patient has been reported to be “demented,” but paper prints of her cranial CT shown to the authors ruled out calcifications. It is therefore likely that the PSEN1 mutation was transmitted from the index patient’s mother but the SLC20A2 mutation from the father.
留言 (0)