
記住我
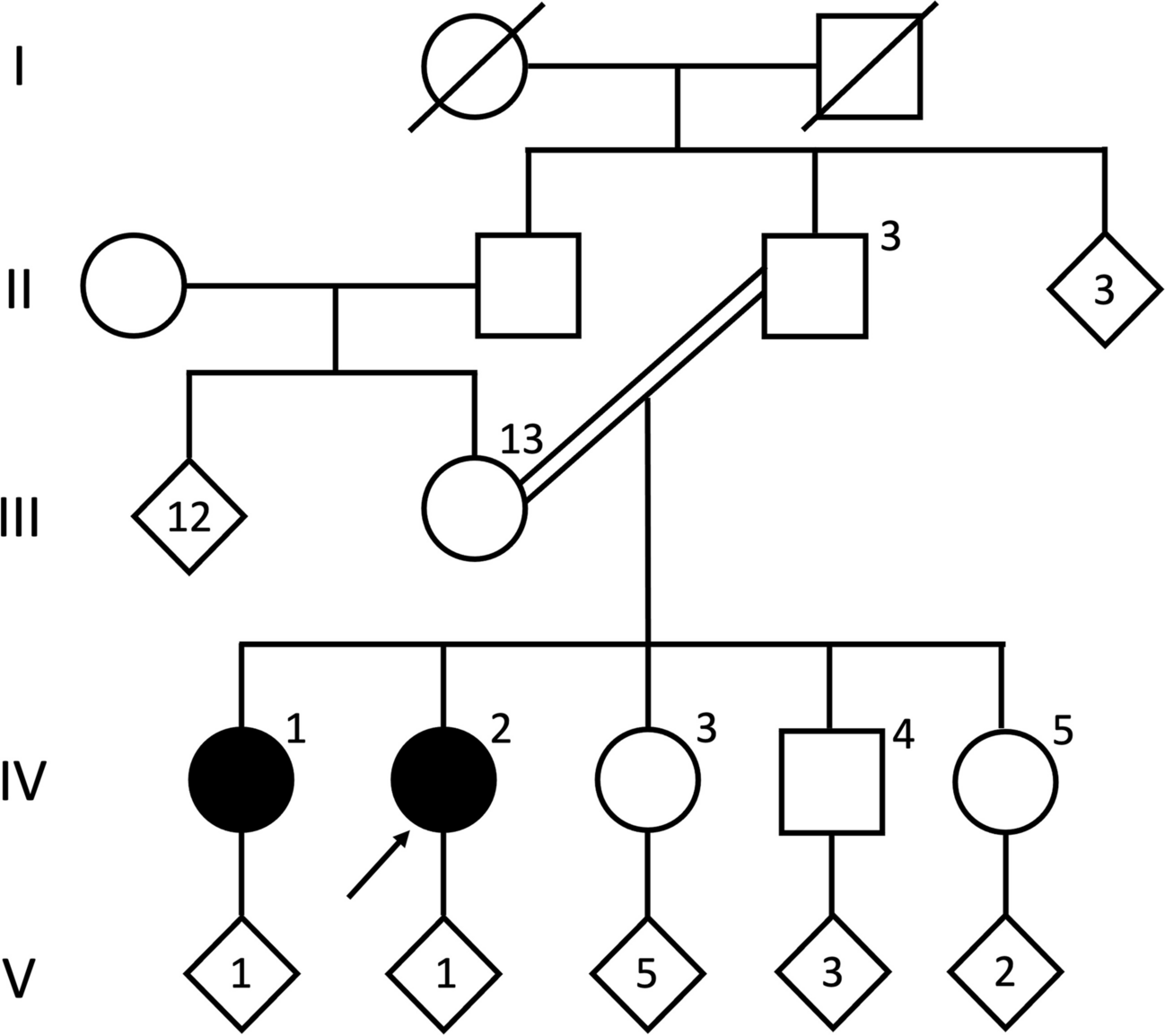
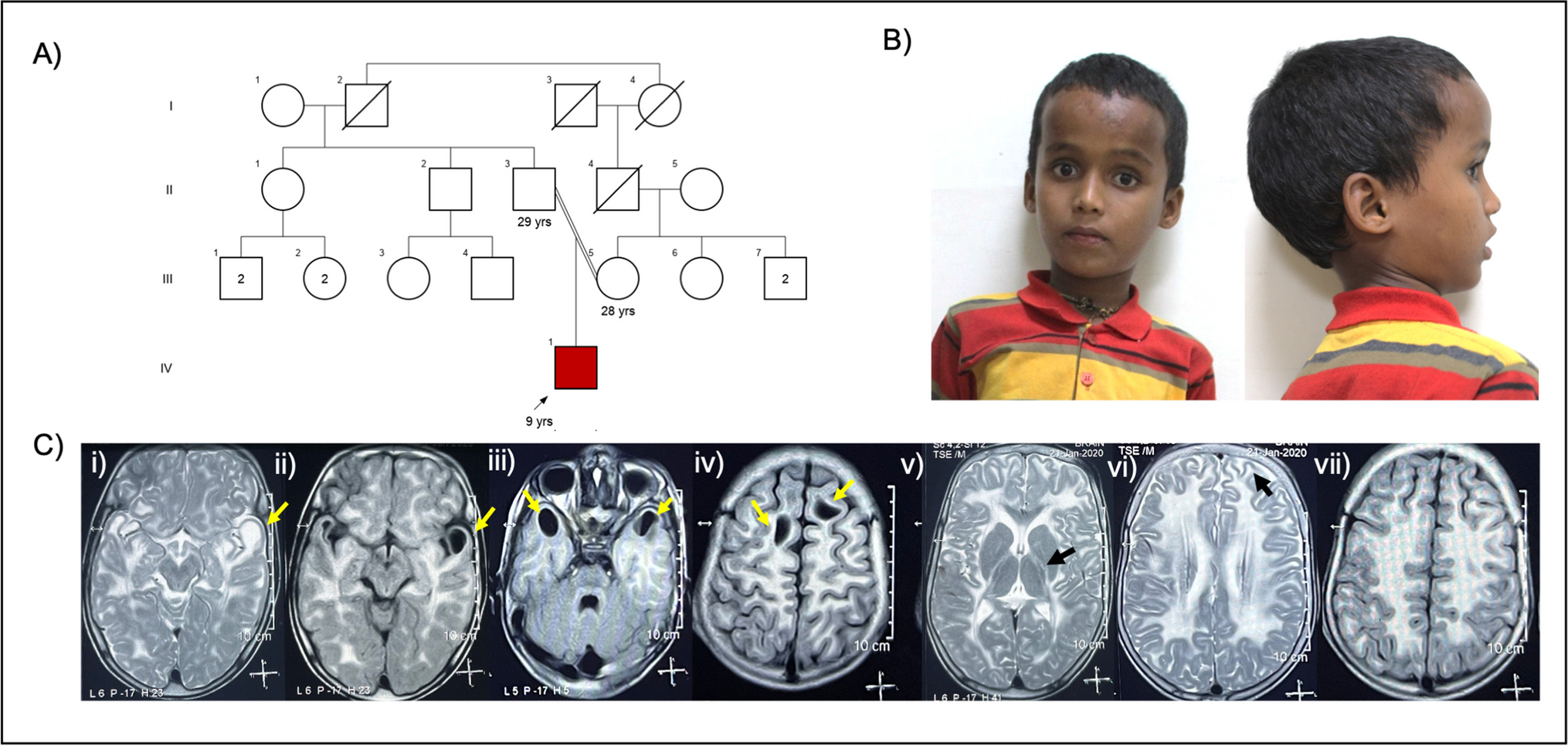

We describe a family consisting of a 35-year-old mother (individual II in Fig. 1), a 36-year-old father (individual II in Fig. 1), and their two children. The elder daughter (individual III in Fig. 1) was 1 year old and her brother (individual III in Fig. 1) was 2 years old at first examination. Both children presenting with moderate-to-severe developmental delay (DD) and intellectual disability (ID), another important feature is epilepsy. The mother (individual II in Fig. 1) and father were normal. The mother has two sisters and one brother (individuals II in Fig. 1), who had their children had a normal development. The father has one sister and one brother (individuals II in Fig. 1), and their children had normal development (Fig. 2).
Fig. 1
Pedigree representing the familial segregation of the 1p36.3 duplication in the family
Fig. 2
A, B The siblings’ hallux valgus and flat feet. C, D The siblings’ sparse eyebrows, sparse eyebrows, hypertelorism, ptosis, and strabismus. E Younger brother’s eyelid myoclonus. F Younger brother’s mild bitemporal narrowing and sloping forehead
Clinical examinationProbandThe first child of this family, a young girl, was referred to moderate-to-severe developmental delay (DD) and intellectual disability (ID). She was born at 39 weeks of gestation, denied history of hypoxia and trauma, birth weight, height, and OFC were normal, and poor feeding in the neonatal period. She was able to sit after 1 year of age and could not walk without support at the age of 12 months. Her language is also delayed, consisting of a few simple words at the age of 18 months. Her medical history was marked by eyelid myoclonus with absence of epilepsy (Jeavons syndrome), treated effectively with sodium valproate since the age of 2. EEG (Figs. 3, 4, 5, and 6): widespread 2.5–3.5 Hz spikes and spike slow complex wave, eye closure sensitivity, and photosensitivity. Wechsler Intelligence Scale score is 72. MRI showed bilateral hippocampal sclerosis. She has an unusual behavior with some autistic signs: she is motionless and impassive with poor interactions and reduced interest in other people or toys. The child has dysmorphic features, including mild bitemporal narrowing and sloping forehead, sparse eyebrows, hypertelorism, ptosis, strabismus, infraorbital creases, wide nasal bridge with bulbous nasal tip, dystaxia, hallux valgus, and flat feet (Fig. 2).
Fig. 3
Eyelid myoclonus with absence in the sister at the age of 5 years, with rapid blinking during play with daze and slight loss of consciousness lasting approximately 5 s in the waking period, and spike-and-slow wave bursts of 2.5–3.5 Hz visible in the full EEG conduction (synchronized with action termination)
Fig. 4
Sister 6 years old with multiple eyelid myoclonus episodes, EEG, extensive medium–high wave amplitude 3–3.5 Hz spike-slow complex wave burst for 1 s
Fig. 5
Open-close eyes test. EEG, eye closure sensitivity for 3 s
Fig. 6
Intermittent photic stimulation (IPS). EEG, extensive spike-slow complex burst
Proband’s younger brotherThe second child of the family, a boy, was born at term after an uneventful pregnancy. Birth weight, height, and OFC were normal. His medical history was marked by eyelid myoclonus with absence of epilepsy (Jeavons syndrome), and treated effectively with sodium valproate since the age of 4. Like his elder brother, she has a mild developmental delay. At physical examination, mild bitemporal narrowing and sloping forehead, sparse eyebrows, hypertelorism, ptosis, strabismus, infraorbital creases, wide nasal bridge with bulbous nasal tip, dystaxia, hallux valgus, and flat feet were noted (Fig. 2).
Family exome sequencing resultsMaterials and methodsGenomic DNA from peripheral blood was extracted using the Whole Blood Genomic DNA Extraction Kit (Beijing Makino Medical Laboratory Institute, Beijing, China) according to the standard operating procedure (SOP). The DNA library was generated by polymerase chain reaction (PCR) using the KAPA Library Preparation Kit (KAPA Biosystems, Boston, USA) according to the SOP. The whole exome was captured using the SureSelect Human All Exon Kit V6 (Agilent Technologies, CA, USA). The target region was sequenced in high throughput using the NovaSeq 6000 platform (Illumina, CA, USA). CNV-sequencing was performed to confirm the pathogenic mutations detected by WES.
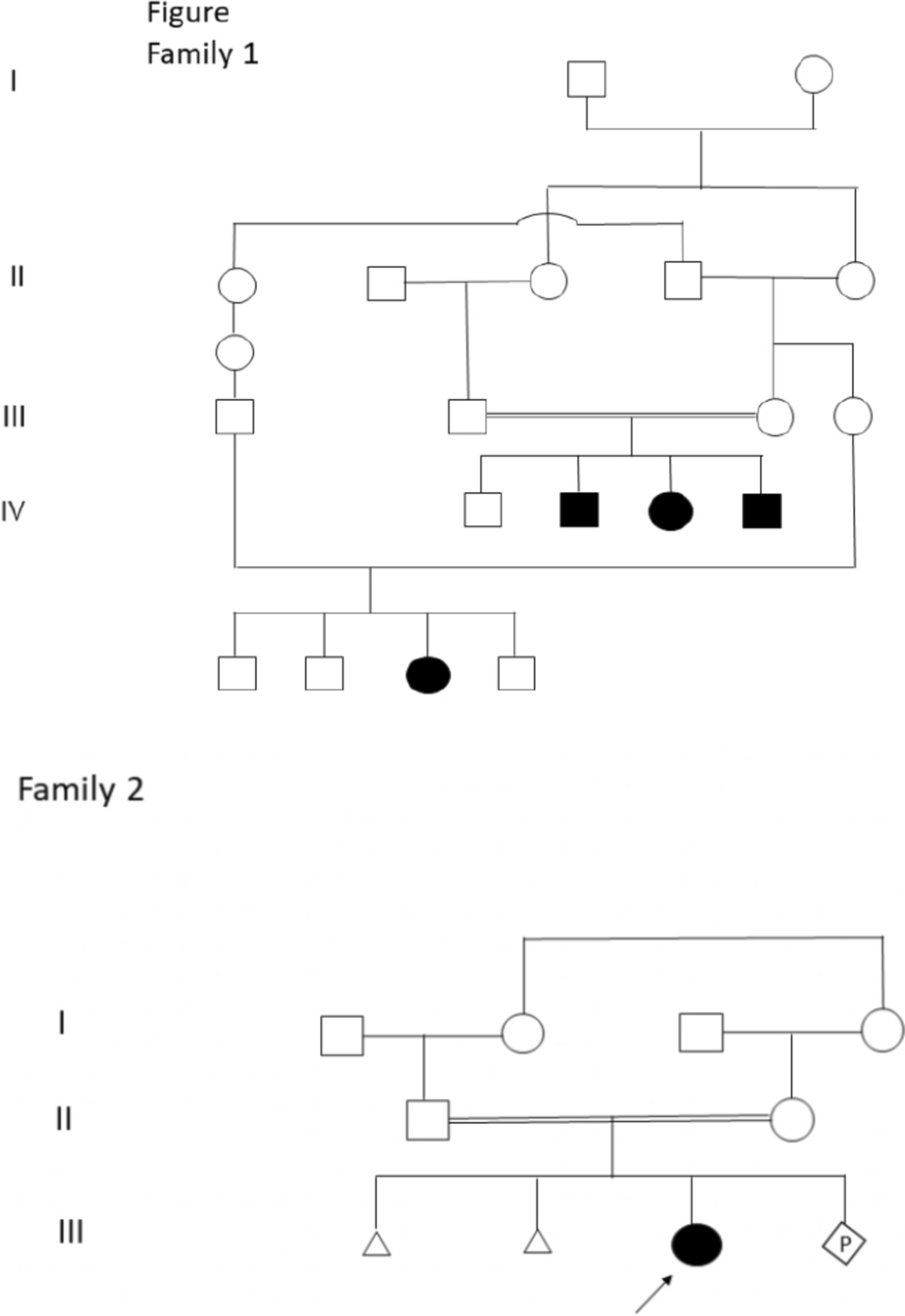
ResultsThe siblings were tested for the same mutation: seq[hg19]dup(1)(p36.33p36.32) chrl: g.621095–3816818dup. The younger brother was sick and also carried the duplication mutation in this segment. However, the variant was not detected in the blood samples of the patient's parents (Fig. 7). The presence of gonadal mosaicism in one of the parents was highly suspected.
Fig. 7
Family whole exon detection results: 20C242115 sister, 20C242116 father, 20C242117 mother, and 20C242118 brother
There were 73 protein-coding genes in the CNV (score = 0.9) (Fig. 8). On the basis of these considerations, we are concerned that the evidence is insufficient for pathogenicity. We searched the DECIPHER database for cases of shorter CNV that were covered by the duplication found in this study. This duplication is associated with 1p36.3 microduplication syndrome, which is consistent; this duplication is associated with 1p36.3 microduplication syndrome, which is consistent with the clinical manifestations of two patients (Table 1). The patients’ genetic information was not applicable and phenotypes were, but consistent with that described in similar cases (score = 0.1), so the ACMG score was pathogenic (P). And we wanted to define the possible pathogenic gene.
Fig. 8
Initial assessment of genomic content
Table 1 Clinical findings in published 1p36.3 duplication carrier patients whose CNV shorter and covered by duplicationsIdentifcation of variants in GNB1In addition, we found that GNB1 may be a repeat intolerance gene (pTriplo score = 1.0); both LoF and DN effects have been described for patients carrying pathogenic GNB1 variants, which would be in line with the hypothesis that this gene has a strong contribution to the phenotypes presented for the siblings.

留言 (0)