1. IntroductionZika virus (ZIKV) belongs to the family Flaviviridae and contains a 11 kb long positive single stranded (ss) RNA genome encoding 3 structural and 7 non-structural proteins surrounded by a nucleocapsid, made from viral capsid protein, enveloped in a host-derived lipid bilayer [
1,
2]. First isolated from non-human primates in 1947, ZIKV outbreaks date back to 2013 [
2,
3]. 2016 marked the peak number of cases in the US according to the CDC [
4]. WHO reported 80 countries with cases of ZIKV transmission as of Feb 2022 [
5]. ZIKV transmission is primarily by Aedes aegyptii mosquitoes [
6]. ZIKV also can be transmitted in utero or by sexual and transfusion methods [
5,
7,
8,
9,
10]. However, these epidemiological results are far from accurate because 80% of infections are asymptomatic or non-specific, lack of sufficient routine surveillance, and cross-reactivity with other flaviviruses like Dengue virus (DENV) [
4,
5,
7]. Some of the symptoms associated with ZIKV infection include acute fever, conjunctivitis, muscle and joint pain, headache and arthralgia [
11]. ZIKV pathology includes a multitude of neurological conditions such as Guillain-Barré Syndrome (GBS) and chronic inflammatory demyelinating polyneuropathies (CIDP) of the peripheral nervous system (PNS) and myelitis, acute disseminated encephalomyelitis (ADEM) and meningoencephalitis of the central nervous system (CNS) [
12,
13,
14,
15,
16,
17]. Other pathologies include acute onset chronic inflammatory demyelinating polyneuropathies, demyelination, axonal injury and other nervous system related conditions [
12,
18,
19]. Some of the symptoms associated with these conditions include mild weakness to severe acute flaccid paralysis in the case of GBS [
20]. Inflammation of the spinal cord and sensory-autonomic impairment has been associated with ZIKV cases of myelitis [
21,
22,
23]. Inflammation of the brain, meninges, headaches, fever, seizures and aphasia in the case of meningoencephalitis, and a chronic progressive symmetric weakness in proximal and distal muscles in the case of CIDP have also been associated with ZIKV infections [
18,
22,
24,
25]. ZIKV infects astrocytes, oligodendrocytes, microglia, and neural progenitor cells, of which astrocytes form the largest cell population [
26,
27,
28]. In utero studies in ZIKV-infected newborns associate intracranial calcifications, microcephaly, and cortical thinning and blindness with ZIKV infection [
29,
30,
31]. Moreover, ZIKV also damages the blood–brain barrier (BBB) integrity via the increased production of inflammatory molecules leading towards viral persistence and replication in the CNS [
32,
33,
34]. Choroid plexus and meninges are also prone to ZIKV infection which provides an additional pathway for ZIKV into the brain parenchyma [
35,
36]. Drugs like azithromycin have been tested as inhibitors of ZIKV infection but no vaccines or treatments are currently available [
37].Astrocytes perform numerous functions. They behave as living scaffolds for neurons, promoting neurogeneration [
38]. Astrogliosis disruption can damage glial scar formation and maintenance of BBB processes [
39,
40]. Astrocytes also homeostatically control glutamate, lactate and Ca++ and K+ ions as part of their regulation of energy metabolism, oxidative stress and synaptic control [
33,
41,
42]. This homeostatic control is crucial for normal muscle movement and behavioral control and if this homeostatic control of these ions does not exist in the CNS, it can result in inflammation, neurotoxicity and Huntington disease related implications [
39,
40,
43,
44]. Astrocytes are among the first cells to be infected by ZIKV.Numerous mass spectrometry studies have been conducted in numerous cell types to examine host protein modifications after ZIKV infection. A label-free approach using LC-MS/MS in microcephalic fetuses found patterns of dysregulations in DNA damage repair response and mRNA translation to be linked with ZIKV infection [
45]. Another TMT 10-plex study on placental tissues associated ZIKV infection with placental integrity compromise [
46]. Mass spectroscopy on 124 serum samples identified fibrinogen alpha platelet factor 4 variant 1 (PF4V1) as a key difference in ZIKV and DENV infections in 62 patients [
47]. ZIKV protein interaction mapping in HEK293T cells highlighted potential novel antiviral drug avenues against ZIKV infection [
48]. LC-MS/MS studies in human fetal neural progenitor cells and on astrocytic cells, respectively, identified dysregulations in cell proliferation, differentiation and migration and neural cell adhesion molecule (NCAM1) as a receptor for ZIKV infection [
26,
49,
50]. iPSC-derived astrocytic cells were also studied and linked ZIKV infection to DNA breakage and reactive oxygen species imbalance in the cells [
51,
52]. RNA approaches to understanding ZIKV infection have also yielded promising results. An RNA-seq study with mouse primary astrocytes revealed pathways related to neuron development, tight junction formation and astrocyte projection to be impacted by ZIKV infection [
53]. We have previously used SOMAScan, an aptamer-based proteomic tool, to delineate astrocytic responses to ZIKV infection [
54]. The SOMAScan platform uses slow off-rate modified aptamers (SOMAmers) that bind to proteins in their native state and are measured using DNA microarray chips [
55]. Here, we used TMT 6-plex, coupled with LC-MS/MS to identify approximately 8000 astrocytic proteins. We also knocked down several of the identified dysregulated proteins and found that KD of several, including major histocompatibility complex class IA (HLA-A), heat shock protein family A member 5 (HSPA5), insulin like growth factor binding protein 5 (IGFBP5) and proteosome 20S subunit alpha 2 (PSMA2) led to increased ZIKV NS1 protein production and ZIKV titers, which suggests each of these proteins normally restrict ZIKV growth in astrocytic cells. 2. Materials and Methods 2.1. Cells Human glioblastoma astrocytoma (U251) cells [U251 MG (previously known as U-373 MG; European Collection of Authenticated Cell Cultures—ECACC 09063001, Sweden)] were grown in Dulbecco’s modified Eagle’s/Nutrient Mixture F-12 (DMEM/F-12) media completed with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 1× non-essential amino acids and 1× sodium pyruvate at 37 °C in 5% CO2. Cells were trypsinized every 2–3 days for passage. Viral titers were determined using plaque assays in Vero cells, as described [
54]. 2.2. Viral Infections
Zika virus (ZIKV), Asian strain, was a gift from Dr. David Safronetz, Chief of Special Pathogens, National Microbiology Laboratory, Public Health Agency of Canada, Winnipeg, MB, Canada. ZIKV was grown in Vero cells in 1× DMEM containing 2.5% FBS, 2 mM L-glutamine, 2× gentamicin, 2× Amphotericin B, 1× non-essential amino acids and 1× sodium pyruvate at 37 °C in 5% CO2. Stock virus was prepared by low multiplicity infection (MOI = 0.001) and viral supernatants harvested at 3–5 days post-infection, supplemented with FBS to 20% and aliquots frozen at −80 °C until used. For experimental infection, 60–70% confluent U251 cells in 10 cm dishes or in 6-well plates were infected at an MOI of 3 to ensure >95% cells were infected at time 0. Virus was adsorbed at 37 °C in 5% CO2 for 2 h with frequent rocking. After 2 h, cells were overlayed with 1× DMEM/F12 media supplemented with 2.5% FBS (with 2 mM L-glutamine, non-essential amino acids and sodium pyruvate, 2× gentamicin and 2× amphotericin B). Parallel mock-infected samples also were prepared. Cells were scraped and the supernatants harvested at 12, 24 and 48 h post infection (hpi) and stored at −80 °C.
2.3. Plaque Assays
Plaque assays were performed in Vero cells grown in 12-well plates to 80–90% confluency. Dilutions of viral samples were prepared from 10−1 to 10−6 in gel saline (137 mM NaCl, 0.2 mM CaCl2, 0.8 mM MgCl2, 19 mM HBO3, 0.1 mM Na2B4O7, 0.3% (wt/vol) gelatin) and duplicate wells were infected with each dilution. After a 2 h adsorption of 100 µL of each dilution at 37 °C in 5% CO2, a 1:1 ratio of 1.2% Type I agarose (Difco Laboratories, Detroit, MI, USA) and (2× completed Medium 199–Medium 199 with 6% FBS, 4 mM L-glutamine, 10 µg of gentamicin sulfate per mL and 3 µg of amphotericin B per mL) was overlayed onto the cells. Cells were incubated at 37 °C in 5% CO2 for 4 days. Plates were stained with 0.04% neutral red in 1% agar in PBS. The plaques were counted 18–21 h post staining.
2.4. Cell Lysis and Quantification of Proteins
Cell pellets from each of 18 samples (3 replicates each of infected samples at 12, 24, and 48 hpi, and time-matched non-infected mock samples) were lysed using MPER® (Pierce; Rockford, IL, USA) solution supplemented with 1× HALT® protease inhibitor (Pierce; Rockford, IL, USA), centrifuged at 14,000× g for 12 min at 4 °C and the lysate separated from the cellular debris. BCA protein assay (Pierce; Rockford, IL, USA) was used to quantify the amount of protein in each sample using bovine serum albumin (BSA) as a standard.
2.5. Mass SpectrometryAll protein samples were processed using the single-pot solid-phase-enhanced sample preparation (SP3) protocol to remove any detergent and salts from the lysates [
43]. Lysates were first reduced by DTT to break tertiary structures including disulfide linkages, and then alkylated using iodoacetamide to prevent re-oxidation. After reduction and alkylation, the proteins were purified using the SP3 carboxylate modified Sera-MagTM beads. The beads were washed 2× with 70% ethanol then by 100% ACN. After the washing steps, trypsin was added at an enzyme to protein ratio of 1:25 and the proteins were digested into peptides, eluted and labelled with the TMT 6-plex system. Labelled samples were analyzed by liquid chromatography/tandem mass spectrometry (LC -MS/MS). 2.6. siRNA-Mediated Knock Down
For initial screens, 20 μM stock solutions of lyophilized siRNA were prepared by dissolving them in 1× siRNA buffer. 5000 U251 cells in each well of a 96-well plate were treated with 80 nM of each of various SMARTPool On-Target plus siRNA targeting a variety of cellular genes. Scrambled control non-silencing siRNA were used for the negative control treatment. Dharmafect/OptiMEM mixture was used for the transfection. After siRNA treatment, cells were incubated at 37 °C for 48 h, after which their cell viability was measured by WST-1 assay (described below). At 48 h post siRNA treatment (hpt), U251 cells were infected with ZIKV at an MOI of 3 (described above) and the supernatants were collected for viral titer measurements using plaque assays. All the experiments were performed in triplicates.
To knockdown specific genes, 20 µM stock solutions of lyophilized siRNA were prepared by dissolving them in 1× siRNA buffer. Knockdown of HLA-A, HSPA5, IGFBP5 and PSMA2 was performed using the SMART-Pool siRNAs for each at 25 nM. Scrambled siRNA was used as negative control. U251 cells were plated in 6-well plates, grown to 40% confluency and siRNA, solubilized in OptiMEM/Dharmfect, were added. Cells were treated with siRNAs for 48 h. In some cases, a second treatment was performed 24 h after the first siRNA treatment. Afterwards, ZIKV infections were done at an MOI of 3 and supernatants and cell pellets were collected for viral plaque assay and viral protein synthesis at 48 hpi. The experiments were performed in triplicates.
2.7. WST-1 Assay
Cell viability was determined using the WST-1 assay in 96-well plate format. Cells in each well were treated with 8 µL of WST-1 reagent after 48–72 h of siRNA treatment and incubated at 37 °C for 1.5 h. Absorbances at 440 nm and 610 nm were recorded using a plate reader and 610 nm absorbance values of infected samples were subtracted from their corresponding 440 nm absorbance values before being normalized to their respective mock samples. An average of 4–6 replicates per condition was calculated to determine cell viability values.
2.8. Western Blotting
10–20 μg of protein lysates were combined with 10% 2-mercaptoethanol, heated for 5 min at 95 °C, and resolved in 12% sodium dodecyl sulfate polyacrylamide electrophoresis (SDS-PAGE) gels at 100–120 V until the loading dye reached the bottom of the gel. Resolved proteins were transferred to PVDF membranes (Immobilon-P polyvinylidene difluoride membrane (Millipore), Sigma-Aldrich Canada Co., Oakville, ON, Canada). The PVDF membranes were blocked with 5% skim milk (wt/vol in 1× TBST) for 1 h at room temperature or overnight at 4 °C. Primary antibodies were mouse anti-ZIKV NS1 (BioFront Technologies # BF-1225-06, Tallahassee, FL, USA), mouse anti-beta actin (Cell Signaling # 8H10D10, Danvers, MA, USA), rabbit anti-HLA-A (GeneTex # GTX114080 N1C2, Irvine, CA, USA), rabbit anti-IGFBP5 (Cell Signaling #3488, Danvers, MA, USA), mouse anti-HSPA5 (EMD Millipore Corp. MABC6750 clone 4E3, Oakville, ON, Canada), rabbit anti-PSMA2 (Cell Signaling 2455S, Danvers, MA, USA) and rabbit anti-GAPDH antibodies. Secondary antibodies used were goat HRP-conjugated anti-rabbit (Cell Signaling # 7074, Danvers, MA, USA) and goat HRP-conjugated anti-mouse (Cell Signaling # 7076, Danvers, MA, USA). Blots were imaged using ECL Western blotting peroxidase substrate for chemiluminescence. ImageJ was used to quantify the Western bands, and each was then normalized to their respective actin or GAPDH controls.
2.9. Statistical and Bioinformatic AnalysesNumerous peptide sequences were identified by mass spectrometric analyses and proteins were identified and quantified from at least 2 different non-redundant peptides in its sequence. This resulted in expectation values and calculated false discovery values of 0.1%, using the xTandem (
https://www.thegpm.org, accessed on 28 October 2022) peptide identification software. Protein quantity fold changes between infected and each time-matched mock samples were converted to log2 values and significance determined by Students t-test and by Z-score analyses as described [
54]. 4. DiscussionWe used a mass spectrometry-based proteomic approach to understand ZIKV infection in U251 cells, followed by siRNA-mediated knockdown studies to delineate the importance of different host cellular proteins in ZIKV replication and protein synthesis. Most of the proteomic pathway analyses were performed using IPA software to generate top ZIKV dysregulated cellular networks, functions and canonical pathways and they helped explain some of the ZIKV-mediated molecular dysregulations that could potentially lead to broader clinical manifestations. The fold-change cut-off was chosen to be >1.333 and
Table 1). For example, OAS3, which negatively regulates interferon/chemokine responsive genes and modulates innate response against Chikungunya infection [
24,
56,
57], was over-expressed (
Figure 2A), while SERP1, which regulates stress responses and interacts with viral NS4B protein to suppress replication in cases of DENV type 2 infections [
58,
59], was under-expressed (
Figure 2A). Similarly, MBD4, which has a role in DNA methylation, repair and CNS development [
41,
60,
61], and MX2, which is a restriction factor for other viruses including HIV, were over-expressed (
Figure 2B), while WNT2B, which regulates cell growth, adult CNS development and differentiation and innate responses via the CTNNB1 signaling pathway [
62,
63,
64,
65,
66], was under-expressed (
Figure 2B).Among the ZIKV dysregulated cellular functions at 48 h post ZIKV infection, which was the timepoint with the largest number of dysregulated proteins, MAPK1 was predicted to be activated and STAT1/2 inhibited (
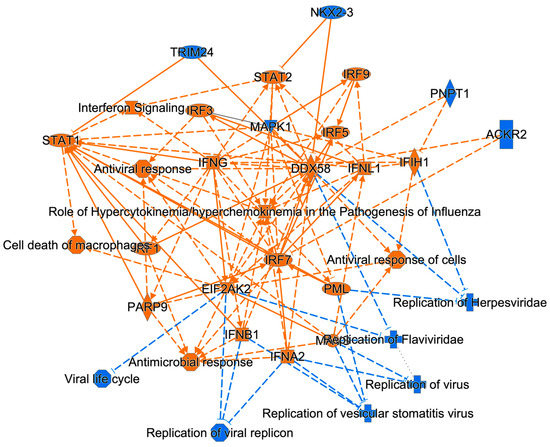
Figure 1). MAPK1 dysregulation induces chorio-retinal atrophy and optic nerve abnormalities in ZIKV infections and STAT1/2 is involved in antiviral responses; in addition, ZIKV NS5 protein-mediated STAT2 degradation modulates type I and III interferon responses [
51,
67,
68,
69]. Other molecules dysregulated by ZIKV among the top ZIKV dysregulated functions at 48 hpi included over-expressed proteins such as STAT1, IFIT2/3 and HLA-A and under-expressed proteins such as LAMP2 (
Figure 3). Many of them have crucial roles in healthy cellular functions and are also involved in other viral infections. STAT1 mediates antiviral type I, II and III interferon responses, regulates ZIKV-mediated induction of cholesterol 25-hydroxylase and interacts with STAT2 for its ZIKV infection regulation via ZIKV NS2A interaction [
68,
70,
71,
72,
73]. IFIT2/3 regulate apoptotic processes, stabilize IFIT1 and promote its binding to viral RNA for translation inhibition [
65,
74,
75]. HLA-A has a role in WNV and DENV infections and disease severity in HIV and SARS-CoV2 infections [
76,
77,
78,
79,
80]. LAMP2 regulates lysosome biogenesis and autophagosome maturation in other viral infections such as African swine fever virus (ASFV) by interacting with ASFV E248R and E199L proteins and DENV infections [
54,
81,
82,
83,
84]. Therefore, their roles in ZIKV infection need to be further examined to help better understand ZIKV modulation of astrocytic functions to aid its infection and replication.Finally, several ZIKV-dysregulated canonical pathways including the Th1 pathway, role of pattern recognition receptors and interferon signaling highlight the role of ZIKV in neuroinflammation and CNS immune modulation as astrocytes are known to be involved in CNS tissue repair, inflammation, NF-kB pathway and MAPK pathways (
Figure 4) [
85,
86]. Moreover, cognitive functions, synaptic plasticity and DNA and RNA viral load control via Type I interferon receptor (IFNAR) signaling are among the other astrocytic functions potentially dysregulated by ZIKV [
85,
87,
88]. We had previously used the SOMAScan platform and identified ZIKV-induced U251 proteomic dysregulations [
54]. Here, we combined the results from both studies, identified 50 proteins dysregulated at least 2.5-fold, either upwards or downwards, and ascertained what effects KD of each of these genes would have (
Figure 6). Proteins like APOBEC3D and HLA-A not only increased ZIKV titers upon being KD but also cell viability (
Figure 5). From the list of siRNA targets, APOBEC3D, HLA-A and IGFBP5 siRNA KD were among the few that increased ZIKV titers while MBD4, TIMP and TNC KD decreased them (
Figure 7). APOBEC3D is a cytidine deaminase and inhibits other viruses such as HIV-1 and human cytomegaloviruses [
89,
90,
91]. Unfortunately, the APOBEC3D antibody we tested did not work, so it was not followed up with. HLA-A class I & II have roles in WNV and secondary DENV infections, and in HIV and SARS-CoV2 disease severity [
76,
77,
78,
79,
80]. IGFBP5 interacts with heparan sulfate proteoglycans (HSGs) and cell-surface matrix glycoproteins, which are receptors for HIV tat, HSV 1&2 and DENV [
92]. PSMA2 and HSPA5 were included because we have found they are also involved in viral replication. PSMA2 is crucial for 20S proteosome complex assembly, degradation of damaged proteins and is involved in influenza-mediated escape from viral clearance via inhibition of NRF2-regulated oxidative stress response while HSPA5 facilitates binding, entry and protein folding for many viruses including Ebola, BDV, MERS-CoV, SARS-CoV2, DENV E and HBV [
93,
94,
95,
96,
97]. After determining the concentration of siRNA that resulted in a successful KD by 48 hpi, with at least 60% cell viability, U251 cells were KD using siRNAs targeting PSMA2, HSPA5, HLA-A or IGFBP5 (
Figure 6 and
Figure 7). This resulted in an increase in ZIKV titers, highlighting their role as potential restriction factors in U251 cells (
Figure 8). HLA-A and IGFBP5 had contrasting results. ZIKV increased HLA-A expression and reduced IGFBP5 expression but KD of each increased ZIKV titers (
Figure 8). This highlights their differing roles in ZIKV replication. IGFBP5, an insulin like growth factor binding protein, is an IGF signaling regulator and is crucial for cell proliferation, growth and survival. Therefore, ZIKV decreasing its expression modulates growth and leads towards increased cell death as we reported previously [
54]. In addition, IGFBP5 is an activator of PI3K/AKT and MAPK pathways which in turn are utilized by other viruses such as Ebola virus for cell entry [
98]. Hence, later upon its KD, it could increase ZIKV efficiency to establish infection and replicate faster in U251 cells. However, HLA-A is a major histocompatibility complex antigen, ubiquitously expressed in nearly all nucleated cells with its role in endogenous peptide presentation to CD8+ T cells. Thus, increase in HLA-A in the presence of ZIKV could be a host response to viral infection and KD of it circumvents this response allowing faster replication (
Figure 8). This highlights how KD of genes, either up- or down-regulated by the virus, can still exert differing effects on viral titers depending on the function and thus further studies are warranted.Interestingly, ZIKV also restored HLA-A expression over time in HLA-A KD cells, highlighting potential cross-talk between HLA-A and ZIKV proteins (
Figure 9 and
Figure 10). This is interesting because HLA-A expression in astrocytes is mostly in cells confined to CNS lesions [
99,
100]. Moreover, HLA-A also has roles in antigen presenting cells (APC). Therefore, it will be interesting to look at how HLA-A directs ZIKV proteins, if at all, to the surface for antigen presentation [
101,
102]. HLA-A alleles are also associated with Guillain-Barré syndrome (GBS) in different populations [
103,
104,
105]. GBS is a rare neurological disorder in which the body’s immune system attacks part of its peripheral nervous system [
106]. In 2020, the HLA-A33 allele was found in a SARS-CoV2 induced GBS patient [
107]. HLA-A also is associated with acute inflammatory demyelinating polyradiculoneuropathy (AIDP) [
108]. Since limited research on HLA-A’s function in nervous system pathologies exist, contrasting associations have been shown between HLA-A and GBS [
109,
110]. In Iraqi patients with GBS, decreased HLA-A:0101 frequency was found in 2016 while in 2014, in GBS patients from East Coast of Australia, HLA ligands were found to be more prevalent [
109,
110]. Interactions between HLA-A and killer immunoglobulin-like receptors (KIRs) have been associated with GBS and Multiple Sclerosis (MS) patients as either risk or protective factors [
109]. In 1998, HLA types were found to be associated with GBS onset in Japanese patients [
111]. HLA-A role has also been shown to be important in Schwann cells that act as facultative APCs in peripheral nervous system and increase HLA Class I expression during GBS [
112]. Therefore, additional work is warranted to understand the role of HLA-A in ZIKV infections. Finally, HSPA5 KD also increased ZIKV NS1 expression (
Figure 10). Since HSPA5 interacts with the ZIKV envelope, regulated unfolded protein response, and alters the ER environment [
83,
113,
114,
115], its role in flaviviral infection also needs to be further examined.Numerous proteomic and transcriptomic studies have been conducted to explain ZIKV infection in microcephalic fetuses, primary human fetal neural progenitor cells, serum samples, placental tissues, and astrocyte-derived cell lines [
45,
46,
47,
49]. This study identified numerous cellular proteins, some of which were similar to previous studies while others were novel. Among the similarities, fibronectin was found to be downregulated by ZIKV in U251 cells in this study. Our previous SOMAScan proteomic study also implicated ZIKV-induced damage to placental integrity [
45,
54]. Cytokines and chemokines such as IL-6 were identified as downregulated while IL-8 and CCL5 were upregulated in this study (
Table 2) and in our previous study [
54]. They were also found to be upregulated in ZIKV-infected human brain cortical astrocytes [
116]. RNA-seq studies done in mouse primary astrocytes revealed common functions such as neuron development, brain development and neuromuscular diseases to be dysregulated by ZIKV [
53], similar to the current study (
Figure 3). Insulin like growth factor responses were also implicated in ZIKV infection by Shereen et al. [
53], highlighting the potential role of IGFBP5 in ZIKV infection. Therefore, further studies to explore it are warranted. EDNRB also was one of the genes that was identified as down-regulated at both the mRNA and protein levels, respectively [
53] and at the protein level in the current study (
Table 2). An orthogonal study in 2018 on ZIKV infection in human neural progenitor neuronal cell line SK-N-BE2 also identified markers involved in similar cellular functions and processes as were identified in this study, including cell growth, cell cycle, cell death, NS development and function, and neurological diseases (
Figure 2 and
Figure 3) [
117]. In addition, molecules involved in PI3K/AKT and ERK/MAPK pathways were found to be implicated in ZIKV infection in both this study (
Figure 2 and
Figure 3) and the orthogonal study [
54,
117]. In 2021, a TMT 10-plex system approach followed by LC-MS/MS on 12 placental samples from 2016 in Puerto Rico revealed cell–cell signaling and neurological disease as among the top dysregulated pathways and functions [
46], similar to some of the findings in our study (
Figure 2 and
Figure 3). In addition to similarities, there are a few differences between this study and the previously published omics studies. For example, we used human glioblastoma astrocytoma U251 cells as a model cell line while Scaturro et al., 2018 used human neural progenitor cells, Shereen et al., 2021 used mouse primary astrocytes and Borges-Vélez et al., 2021 used placental samples [
46,
53,
117]. Furthermore, Shereen et al. performed an RNA-seq study while we complemented the LC-MS/MS proteomic study with genetic KD [
53]. Among the novelties in the findings of the current study, antigen presentation was predicted to be upregulated by ZIKV infection in U251 cells at the protein level (
Figure 3) [
49]. This is consistent with the increase in HLA-A expression observed in ZIKV-infected U251 cells and the consequent increase in ZIKV replication and NS1 protein synthesis post HLA-A KD conditions (
Table 2;
Figure 10). Energy production dysregulation was a cellular function differentially identified in this study unlike LC-MS/MS studies done previously on primary neural progenitor cells and human placental samples in 2018 and 2021 [
46,
49]. Another difference between this study and the orthogonal study done by Scaturro et al. is that they focused on ZIKV host protein–protein interactions and phosphoproteomic profiling via affinity purification integrated LC-MS/MS (AP-LC-MS/MS) and on kinase substrate relations/regulatory sites through PhosphitePlus51 resource while this study looked at proteomic impact of ZIKV infection via LC-MS/MS.


Comments (0)