
Remember me
The proband is an 8-year-old female born to non-consanguineous parents from Chuvashia. She was delivered at 37 weeks of gestation with a birth weight of 3160 g (+ 0.66 SD) and length of 52 cm (+ 1.95 SD). The perinatal period was unremarkable with the early years notable only for partial congenital anal atresia and persistent constipation. Her developmental milestones were within expected ranges. Neutropenia (400 cells/µL) was first recorded at 6 months of age. Since 9 months of age, she has had episodes of RALF typically triggered by fever and respiratory infections. Peak serum aspartate aminotransferase and alanine aminotransferase (AST/ALT) levels reached 12,030 and 9,050 U/L, respectively. Coagulopathy, reflected by a prolonged prothrombin time (PT) of 23.7 s and an international normalized ratio (INR) of 1.64, was occasionally observed during episodes. Between episodes her liver enzymes were either within normal ranges or only mildly elevated (e.g., 44/17.5 U/L). Her absolute neutrophil count ranged from 400 to 1500 cells/µL during and between episodes.
She presents with hepatomegaly and imaging signs of liver fibrosis. Cytological examination of the peripheral blood smear showed no significant morphological changes in neutrophils. Bone marrow cytology revealed moderate depletion of the neutrophil lineage without morphological abnormalities and cytogenetic analysis showed a reduced number of mitoses. Immunophenotyping of peripheral blood lymphocytes revealed no abnormalities: CD3 + T cells: 2.09 × 10⁶/mL; CD3 + CD4+ (Th): 1.22 × 10⁶/mL (naive 73%); CD3 + CD8+ (Tc): 0.71 × 10⁶/mL (naive 74.3%); CD19 + B cells: 0.44 × 10⁶/mL (switched 11.3%, CD21lowCD38low 3.9%); CD3–CD16 + CD56 + NK cells: 0.23 × 10⁶/mL. NK cell degranulation was normal.
The proband’s family history is notable: both brothers exhibited a similar clinical presentation with recurrent infection-triggered liver failure, elevated transaminases and neutropenia. One died at 13 months from acute liver failure and the other at 8 years due to multi-organ failure. Prior gene panel testing identified no causative variants.
The proband receives home-based education to reduce infection risk and is under hematology and gastroenterology supervision for neutropenia and recurrent liver failure, showing a good response to granulocyte colony-stimulating factor therapy.
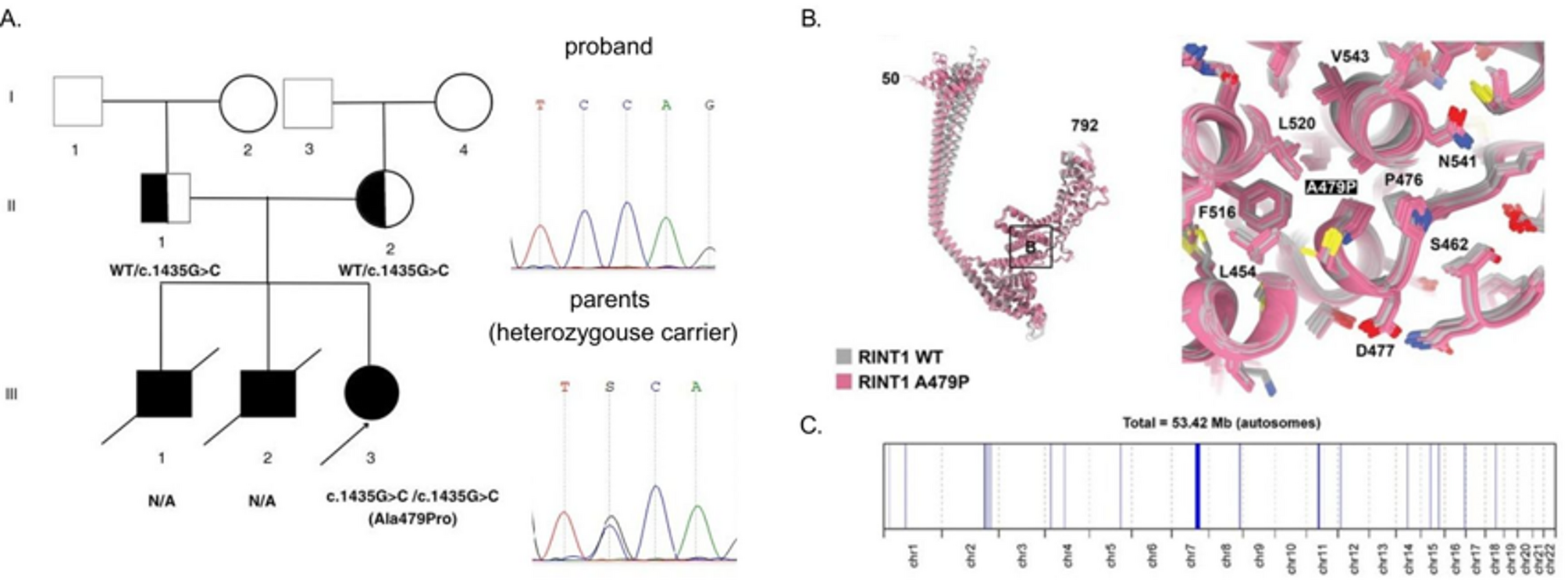
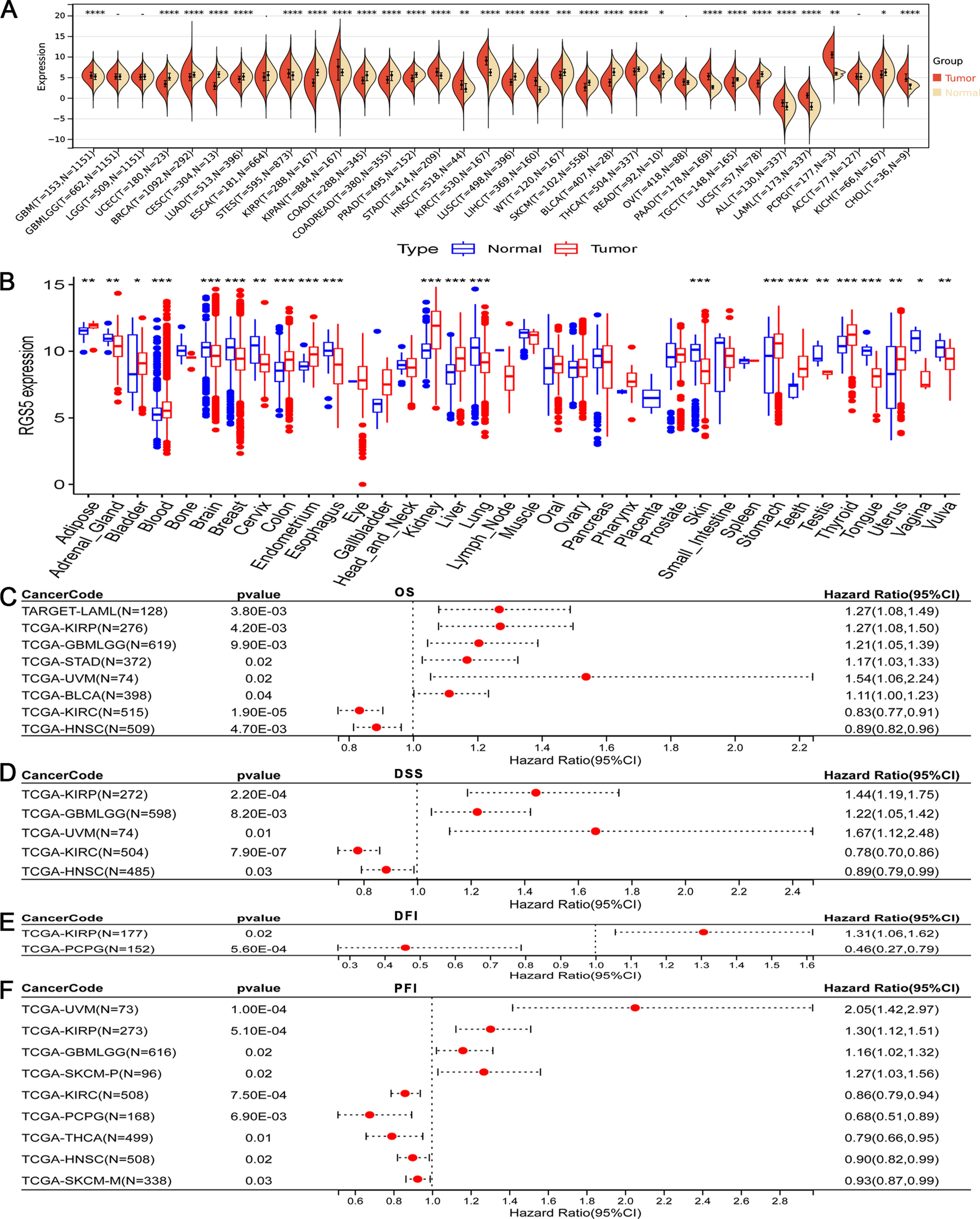
Molecular genetic analysis identified a novel variant, NM_021930.6:c.1435G > C, in a homozygous state in the proband, located in exon 10 of the RINT1 gene. Sanger sequencing confirmed the homozygous state in the proband and a heterozygous state in both parents, as shown in Fig. 1A, B.
Fig. 1
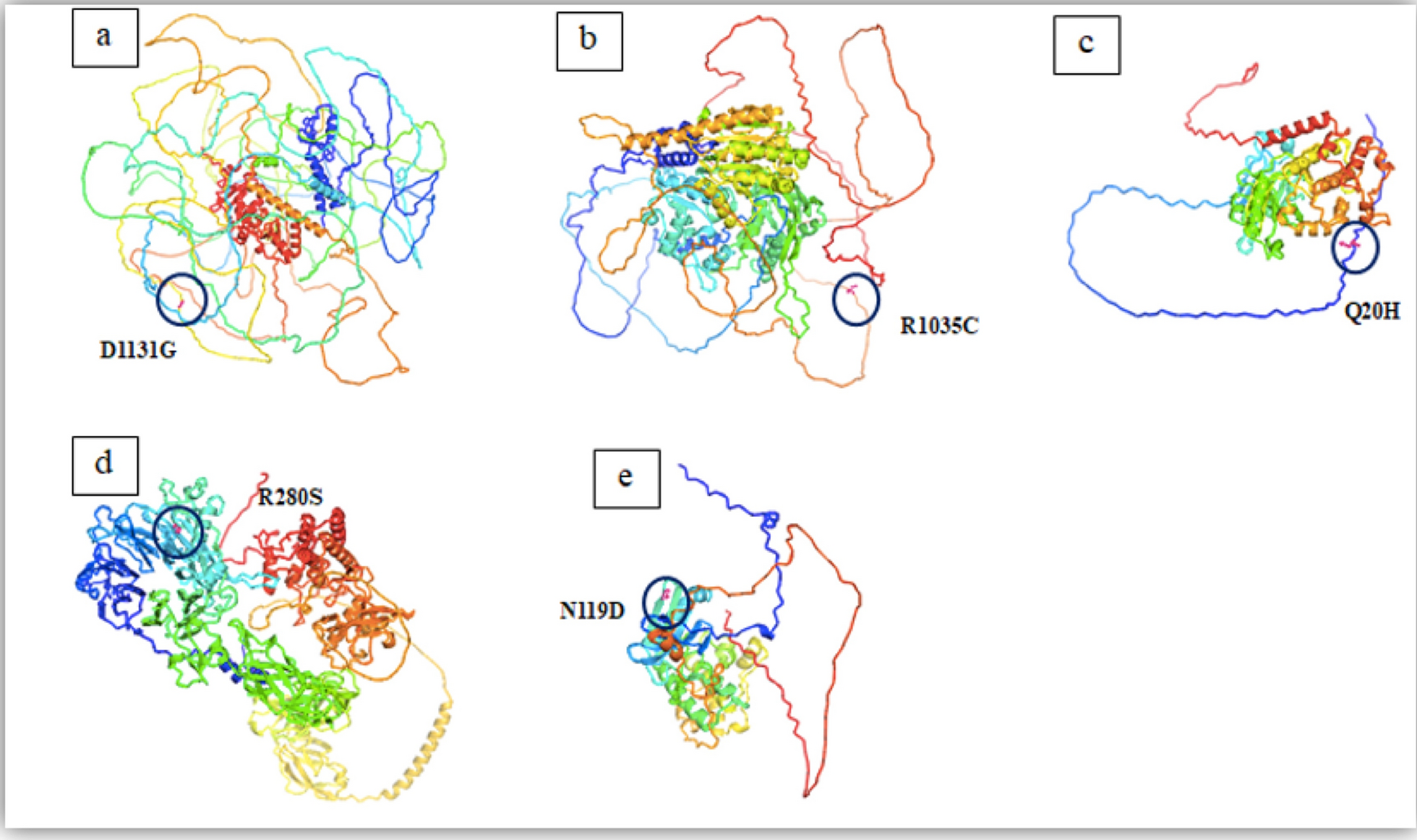
Family pedigree, structural modeling of the RINT1 p.Ala479Pro substitution and mapping of the ROH region. A Pedigree showing the affected proband and siblings with unknown genetic status, and unaffected heterozygous parents. B Superimposed 3D structural models of wild-type RINT1 (gray) and the p.Ala479Pro variant (pink) generated with AlphaFold2. Five top-ranked models are shown for each protein. The Ala-479 to proline substitution within the α-helix of the RINT1/TIP20 domain induces local conformational changes and reduces helix stability by introducing a destabilizing kink. C ROH mapping based on WES data of the proband. Blue lines represent ROH regions distributed across the chromosomes, shown along the X-axis with their respective positions and lengths
This variant results in a missense substitution of the highly conserved amino acid residue p.(Ala479Pro) within the RINT1/TIP20 domain in the RINT1 protein. The identified nucleotide sequence variant is not registered in the control sample of the Genome Aggregation Database (gnomAD v.4.1.0). Most bioinformatic predictors suggest that the variant is likely to be damaging, as shown in the Supplementary. WGS of the proband revealed no additional variants that could explain the clinical presentation.
The functional implications of the identified missense substitution were further investigated through 3D structural modeling of the wild-type (WT) and mutant proteins as shown in Fig. 1, B. The five best Alphafold 2 computed structure models (CSMs) of WT RINT1 (grey) and the five best AF2 CSMs of the A479P variant (pink) are superimposed. Superposition of the best CSMs of WT RINT1 and A479P variant in the proximity to the mutation illustrating moderate local changes in the structure.
The analysis demonstrated that the conserved Ala-479 residue is situated within the alpha-helix of the RINT1/TIP20 domain. Substitution with proline introduces significant structural constraints due to the rigid side-chain ring that incorporates the nitrogen atom, preventing rotation around the N⎯C (alpha) bond. Consequently, proline residues disrupt helix stability by introducing a destabilizing kink and exhibit a low propensity to participate in helix formation [4].
Since the identified variant was detected in a proband in a homozygous state, we conducted detailed genetic counseling for the family to investigate potential consanguinity. The parents, however, were unable to establish a direct genealogical link between their pedigrees. This observation suggests a possible common origin of the identified variant within the Chuvash population, from which the proband’s parents originated. To further explore this, we performed an analysis of ROH using WES data. This analysis revealed an extensive ROH at chromosome 7 spanning 18.5 Mb, with boundaries at chr7:102,605,864–121,125,900 as shown in Fig. 1, C, encompassing the entire RINT1 locus (chr7:105,172,648–105,208,124).
Using Rutgers Map v.2, the physical boundaries of autozygosity regions determined by the WES data were converted into 111.43–126.31 cM (Kosambi) sex-averaged genetic distances, which corresponds to a common ancestor of 6.72 generations ago. Based on the average length of generations in humans of 25 years these estimates suggest the age of the mutation spread of 168.01 years.
Considering all evidence, we classify the variant as likely pathogenic (PM2, PP3, PM1, PP4) and confirm ILFS3 in our proband.
Comments (0)