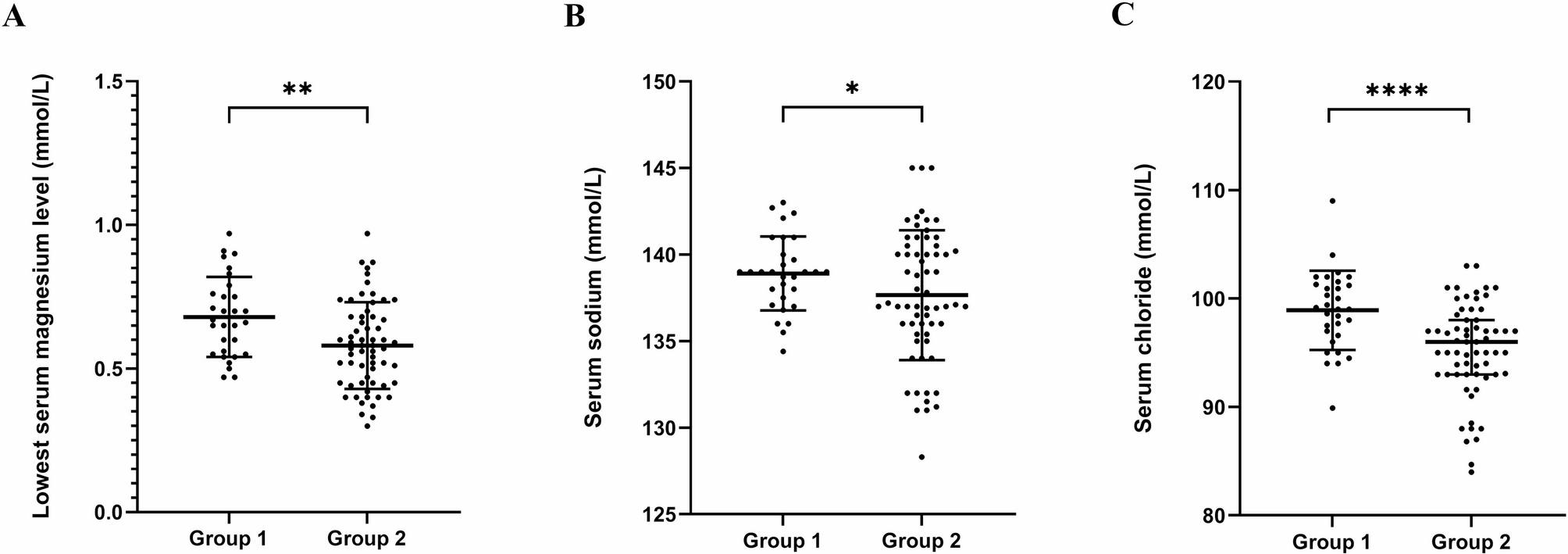
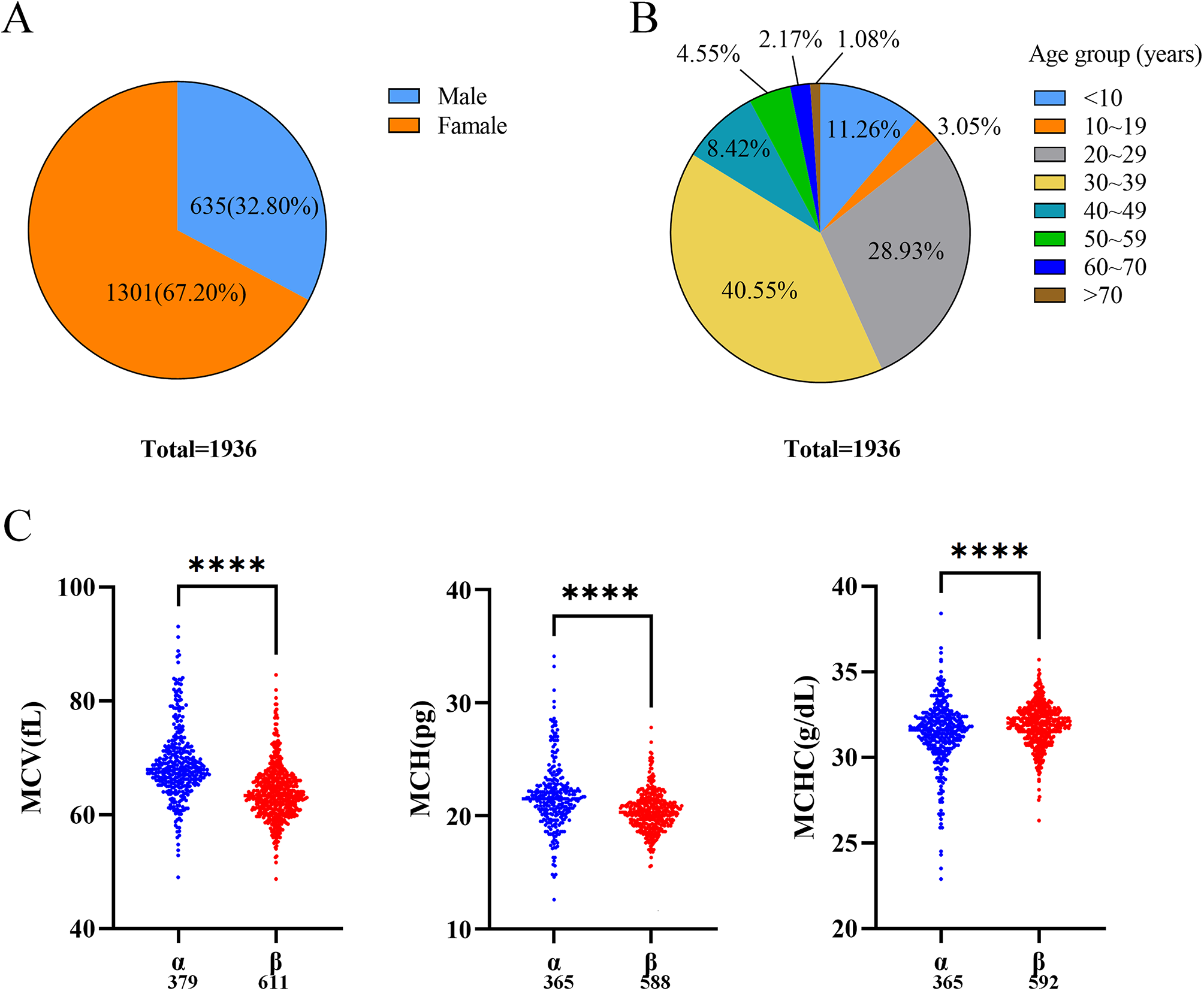
GS often presents insidiously with mild symptoms, leading to frequent misdiagnosis or delayed diagnosis. Typical clinical features include hypokalemia, hypomagnesemia, metabolic alkalosis, hypocalciuria, and activation of the renin-angiotensin-aldosterone system [3, 16]. In this study, 95 GS patients were enrolled, of whom 92 received genetic testing. The results revealed that 76 patients had biallelic variants in SLC12A3, confirming the molecular diagnosis of GS, while 16 patients carried a single variant, with the diagnosis established based on clinical and genetic findings. The remaining 3 patients declined genetic testing and were diagnosed based on typical clinical indicators according to the guideline [1]. The typical clinical indicators of these 19 patients were detailed in Table S4. These enrolled patients had a wide age range (2–52 years), with a median age at diagnosis of 16 years and a balanced gender distribution, which facilitated the creation of age groups (younger group, ≤ 16 years; older group, >16 years) and provided high-quality clinical data for analyzing the phenotypic characteristics and their relationship with age. The majority of patients initially presented with fatigue and sought medical attention only when more obvious symptoms emerged, such as numbness, palpitations, febrile episodes, cramps or muscle weakness. Notably, older patients were more likely to exhibit numbness and palpitations, whereas younger patients commonly presented with febrile episodes, nausea and vomiting. GS should be highly suspected in the presence of persistent unexplained hypokalemia. Further analysis of age and electrolyte levels revealed that older patients had significantly lower serum potassium and magnesium levels compared to younger patients, with both potassium and magnesium levels showing a downward trend as age increases. These findings emphasized the need for long-term electrolyte monitoring and standardized supplementation in GS patients to prevent both under- and over-supplementation.
As a salt-losing tubulopathy, GS treatment primarily focuses on electrolyte supplementation, aiming for serum potassium ≥ 3.0 mmol/L and serum magnesium ≥ 0.6 mmol/L, as recommended by guidelines [1]. The study results indicated that when serum potassium levels were ≥ 3.0 mmol/L, the levels of other electrolytes (magnesium, sodium, and chloride) were generally higher compared to when potassium levels were below target. This suggested that achieving target potassium levels should be prioritized during treatment to maintain electrolyte balance, thereby facilitating disease control. Irregular treatment due to mild symptoms should be avoided. In daily management, electrolyte loss due to excessive sweating, urination, and diarrhea should be prevented, and excessive potassium supplementation should be avoided to reduce the occurrence of side effects such as abdominal pain and diarrhea. Furthermore, the correlation analysis between serum potassium and magnesium levels revealed that elevated serum potassium levels were associated with increased magnesium levels, supporting the notion that magnesium supplementation facilitated potassium absorption [17]. Therefore, GS patients with hypomagnesemia should pay attention to monitoring serum magnesium levels and supplementing magnesium as needed. Additionally, dietary management plays a crucial role [9], and patients are advised to consume foods rich in potassium (e.g., bananas and oranges), magnesium (e.g., nuts and leafy greens) and sodium (e.g., moderate amounts of salt) to support electrolyte balance [18, 19].
Pathogenic variants in the SLC12A3 gene is the molecular standard for diagnosing GS. This study identified 73 SLC12A3 variant sites, including missense, nonsense, frameshift, deletion, insertion, indel, splicing, and intronic variants, with missense variants being the most common, consistent with the previous study [20]. The high-frequency variants (c.179 C > T and c.1456G > A) were consistent with those previously reported in the Chinese population [21, 22], while showing no overlap with the seven high-frequency variants found in European populations (c.938 C > T, c.1108 + 1G > T, c.2221G > C, c.2576T > C, c.2581 C > T, c.2883 + 1G > T, and c.2981G > A) [10], suggesting potential racial differences in gene mutations, which may be attributed to founder effects, population genetic background or sample size. Additionally, six novel variants were identified, with their pathogenicity assessed based on ACMG guidelines, thereby expanding the GS-related variation database and providing valuable reference for genetic counseling.
GS is an autosomal recessive disorder, but only a single heterozygous variant was found in 17.4% of patients, suggesting the likely presence of a second undetected variant. Previous studies have reported that approximately 8–30% of patients carry only a single detectable variant, possibly because the other variant is located in a deep intronic region, involves a large fragment deletion or duplication, or occurs outside the SLC12A3 gene [23, 24]. Therefore, genetic screening should employ advanced techniques, including MLPA, whole-exome sequencing (WES), and whole-genome sequencing (WGS), to identify potential variants and enhance diagnostic accuracy [25]. With informed patient consent, copy-number variation screening (MLPA) is recommended for all patients with single variants in order to more comprehensively clarify the disease and investigate the diversity of genetic variations.
GS is phenotypically and genetically heterogeneous, with phenotype-genotype correlations remaining a research focus. However, varying conclusions have been reported, likely due to sample size limitations. The majority of GS patients in this study were compound heterozygotes, consistent with previous reports [1, 21]. We found that patients in the compound heterozygous group presented a milder degree of hypomagnesemia compared to those in the homozygous and heterozygous groups, but no significant differences were observed in serum potassium, sodium or chloride levels among the three groups. We also found all patients with deletions (5 cases) and indel variants (1 case), as well as the majority of those with frameshift and splicing variants, were diagnosed at a younger age. In contrast, all patients with insertions (3 cases) and most with nonsense or intronic variants were diagnosed at an older age. These findings suggested a potential correlation between age at diagnosis and variant types.
Although we provided a relatively comprehensive description of demographic characteristics, clinical manifestations and serum electrolyte levels, gaps remain in other laboratory parameters, such as urinary electrolytes and plasma renin activity. While the correlation between phenotype and genotype in GS was explored, the limited sample size prevented a full investigation, emphasizing the need for a larger cohort in future studies.
As we enter the age of precision medicine, the understanding of GS has improved, but challenges in diagnosis and management remain. First, comprehensive clinical evaluation of patients must be strengthened. Second, differential diagnosis from Gitelman-like syndrome is crucial, as it shares clinical features with GS and involves multiple causative factors, including genetic (mitochondrial mutations) and acquired (drug-induced, chronic diarrhea and vomiting) components. In addition, fundamental research is needed to investigate the molecular pathogenesis of GS and identify novel therapeutic targets.

Comments (0)