Study Design
This was a randomized, multicenter, double-blind (DB), placebo-controlled, parallel-group phase 3 trial to evaluate the efficacy and safety of sarecycline in Chinese patients aged 9–45 years with moderate-to-severe AV over 12 weeks (W) (DB period). It was followed by an OLFU period to assess the long-term safety and maintenance of sarecycline efficacy over 36W. Total trial duration was 48W.
The trial was conducted at 34 centers in China (Appendix S1 in Electronic Supplementary Material [ESM]). During the DB period, patients were randomized 2:1 with an interactive web response system to receive either sarecycline (approximately 1.5 mg/kg) or placebo as a once-daily oral tablet according to body weight (60 mg for 33–54 kg; 100 mg for 55–84 kg; 150 mg for 85–136 kg) [20] for 12W. During the screening visit, eligibility and washout from prohibited therapies were established. After the baseline visit, patients would visit the clinic every 3W up to W12 to perform efficacy and safety assessments. At the investigator’s discretion, any medication taken for medical reasons was continued at the same dose and conditions during the entire experimental phase of the trial. Other treatments for acne, including topical and medicated cleansers and therapies, were prohibited during the study. Use of systemic antibiotics for indications other than acne were considered on a case-by-case basis.
Patients who responded to treatment at the end of the DB period (≥ 50% reduction from baseline in facial inflammatory lesion counts at W12) were offered to continue in the OLFU for an additional 36W. These patients had their first study visit on the same day the DB period was completed (W12) or within the following 4 months if no other AV treatment was administered during this time. During the OLFU, patients visited the clinic every 12W up to 48W, and treatment was reinitiated for 12W if acne recurred [Investigator’s Global Assessment (IGA) score of ≥ 3, indicating moderate/severe acne]. Trial design is represented in Fig. S1 in ESM. Placebo and sarecycline tablets had the same appearance to ensure blinding to the treatment.
Study protocol was approved by the ethics committees and local regulatory authorities. The trial was conducted according to the principles of the Declaration of Helsinki and the International Council on Harmonization Guidelines for Good Clinical Practice. Written informed consent (including consent to publish the photographs) was provided by enrolled patients or their legal representatives. This study followed the Consolidated Standards of Reporting Trials (CONSORT) reporting guideline.
Patients
Eligible patients were 9–45 years old with a body weight of 33–136 kg, facial AV (20–50 inflammatory lesions [papules, pustules, and nodules], 30–100 non-inflammatory lesions [open and closed comedones], ≤ 2 nodules, and an IGA score of moderate-to-severe). Treatment responders continued in the OLFU if no other treatment had been administered prior to enrollment into this period.
Endpoints
For the DB period, the primary efficacy endpoint was absolute change from baseline (CFB) to W12 in facial inflammatory lesion counts. Secondary endpoints were percent CFB (%CFB) to W12 in facial inflammatory lesion counts and absolute and %CFB to W6 and W9 in facial inflammatory lesion counts. Additional efficacy endpoints included absolute and %CFB to W3 in facial inflammatory lesion counts, percentage of patients with facial IGA success (defined as the proportion of patients with ≥ 2-point decrease from baseline on IGA score and rated as clear [0] or almost clear [1]), absolute and %CFB in facial non-inflammatory lesion counts and non-facial IGA assessment with ≥ 1 point decrease from baseline for the neck, chest and back, by visit. Safety endpoints included number of AEs and serious AEs (SAEs), laboratory parameters, physical examinations and vital signs.
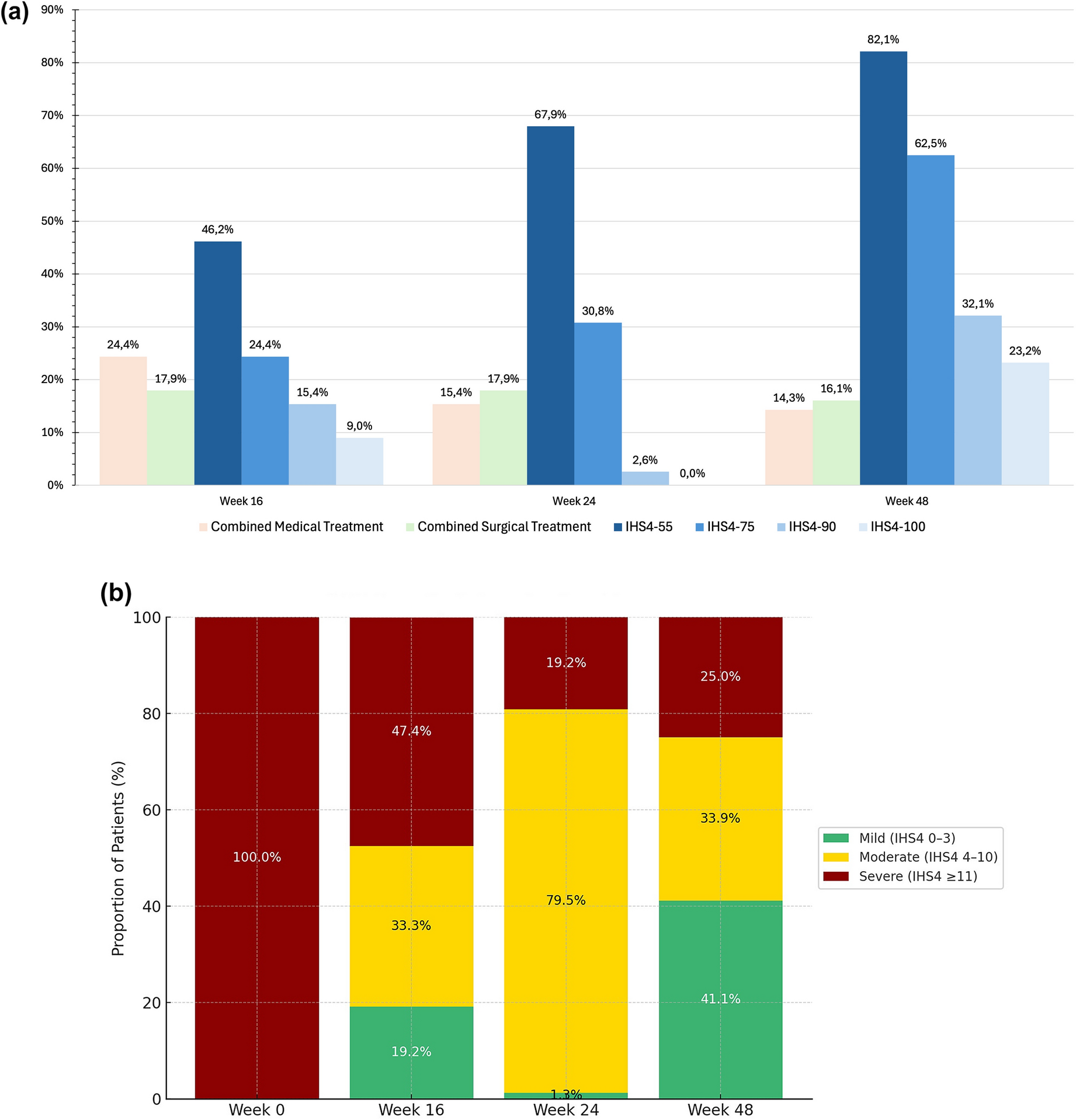
For the OLFU, primary endpoints were number of AEs and CFB in clinical laboratory parameters and vital signs. AEs occurring after treatment had been re-initiated were considered treatment-emergent AEs (TEAEs). Other endpoints included number of recurrences, time to recurrence and efficacy of treatment after recurrence.
Statistical AnalysisDB Period
The sample size of 390 (sarecycline N = 260; placebo N = 130) was determined to provide 90% power at 5% significance level (two-sided) to detect a difference of 4.8 between treatment arms (15.2 for sarecycline and 10.4 for placebo) in the absolute CFB to W12 in facial inflammatory lesions assuming a common standard deviation (SD) of 13.7 [17].
The intent-to-treat (ITT) population was defined as all patients randomized to receive the investigational medicinal product (IMP). Per-protocol (PP) population was defined as patients who completed the DB period, did not have any major protocol deviations that could potentially impact the efficacy and had an overall compliance of ≥ 80%. Analysis of efficacy endpoints in the DB period was performed on the ITT population and the PP population to assess the robustness of the ITT population findings. The DB period was conducted under strict mobility restrictions in China, which made attendance to scheduled visits difficult. The consequent increase in protocol deviations emphasizes the importance of analyzing the efficacy endpoints using the PP population. Safety endpoints were analyzed using the safety population, which was defined as all patients who received at least one dose of the IMP.
Missing values were imputed using multiple imputation (MI) for primary efficacy analyses with the full conditional specification regression method, creating 200 imputations.
Difference between treatment arms was analyzed for each completed dataset using an analysis of covariance (ANCOVA) model, for continuous efficacy endpoints, with the baseline value as a covariate and adjusted by pooled site. For categorical variables, the Cochran-Mantel-Haenszel test adjusted by pooled site was used for the comparison between treatment arms. Homogeneity of treatment effect across pooled sites was analyzed using the Breslow-Day test.
SAS software version 9.4 was used for the statistical analyses. All significance tests were two-sided using 5% significance level. Primary and secondary efficacy endpoints during the DB period were tested with multiplicity adjustment via a fixed sequence testing procedure to control the family-wise type I error rate at α = 0.05 (two-sided).
OLFU Period
Analyses of long-term safety and efficacy maintenance endpoints in the OLFU were performed using descriptive statistics. The baseline used in analyses for the DB period was also the baseline in the OLFU analyses.






Comments (0)