
Remember me

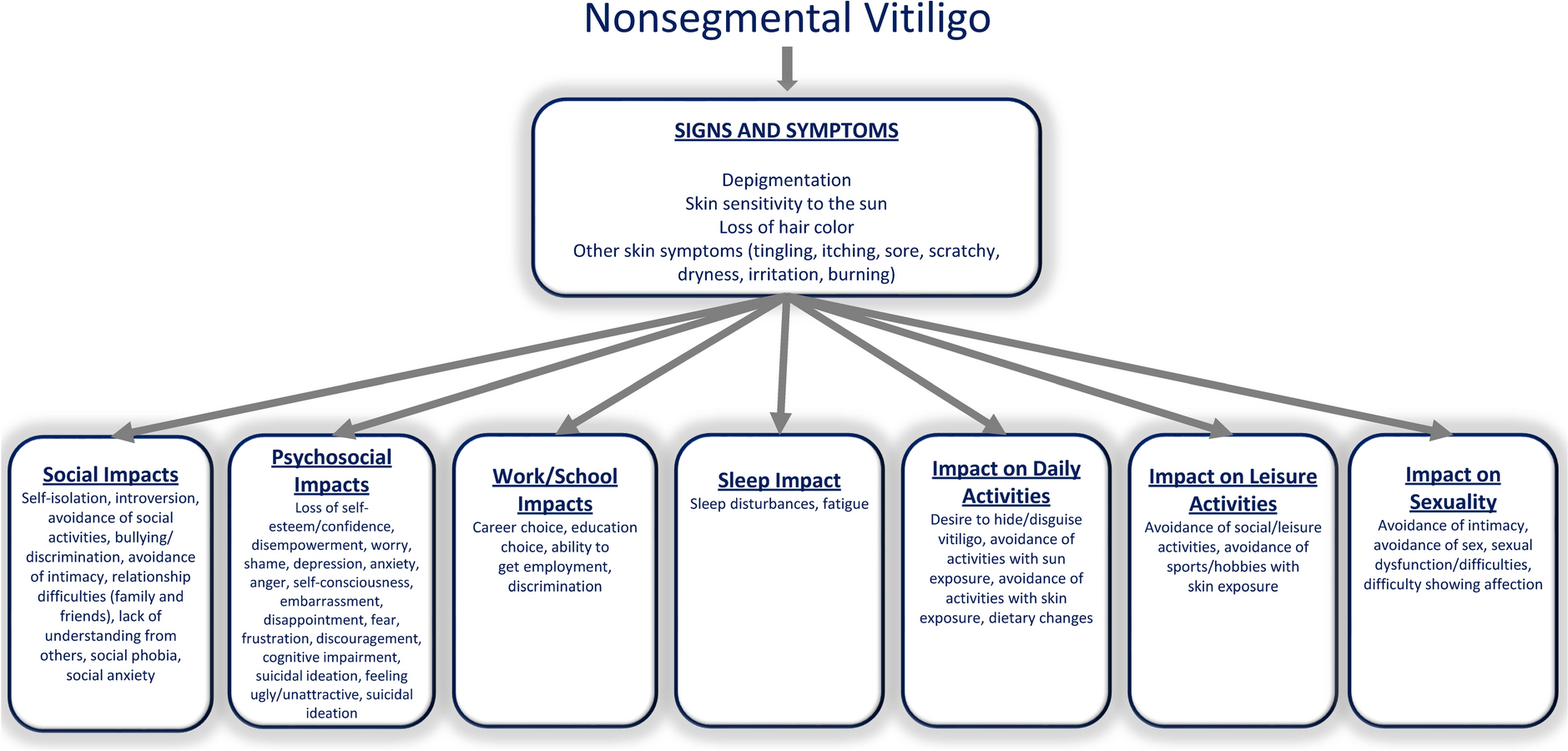




The DAMASK study (NCT05656911) was a randomized, double-blind, placebo-controlled, phase 2a, proof-of concept study consisting of a screening period of up to 28 days, a 12-week intervention period, and a 4-week follow-up period (Fig. 1). This study was conducted in compliance with the International Council for Harmonisation Good Clinical Practice, including the archiving of essential documents. Informed consent was obtained before initiation of any study-related procedures. The study was conducted from December 21, 2022, to January 31, 2024, at 18 sites in 6 countries in Europe (Germany, France, Italy, Poland, Czech Republic, and the UK) and at 3 sites in the US.
Fig. 1
Study design. AD, atopic dermatitis; BID, twice daily; FU, follow-up; EOS, end of study; EOT, end of treatment; S, screening; R, randomization; V, visit
At least 7 days before randomization, patients were required to use a stable amount of topical emollient to moisturize the skin, applied to the whole body BID and continued during the study. Emollients used by the patients before entering the study were to be continued with the same frequency and intensity. During the same 7-day period, patients recorded the daily intensity of peak pruritus using a 0–10 numerical rating scale (NRS) on an electronic handheld device.
Patients were then randomized in a 2:1 ratio to receive oral zabedosertib 120 mg BID or matching placebo. The selected dose of 120 mg BID was the dose that provided a maximum plasma exposure (higher doses of zabedosertib tested in phase 1 did not provide higher exposure levels [6]; it had shown distinct efficacy in human in vivo systemic and topical skin challenges, achieving a skin exposure of approximately 50% of plasma exposure levels [5]. This dose also covered the anticipated therapeutic exposure in various preclinical inflammation models [1, data on file]. Zabedosertib was also effective in reducing the Canine Atopic Dermatitis Extent and Severity Index and itch in two field studies in dogs with canine atopic dermatitis (CAD), a relevant model for AD due to its similarities to its human counterpart [15, data on file].
In the event of a disease flare or AD symptoms, predefined topical rescue medication could be used for several days at the discretion of the investigator. Disease flare was defined as a validated Investigator’s Global Assessment for Atopic Dermatitis (vIGA-AD) score ≥ 3 requiring escalation of treatment confirmed by an investigator, which could not be tolerated by the patient. Before rescue medication was initiated, increasing the frequency of emollient use was encouraged to try to control AD symptoms, and efficacy assessments were performed. The preferred rescue medication included was methylprednisolone aceponate cream or an alternative TCS with similar potency if methylprednisolone aceponate was not available in the patient’s country. If TCS were not tolerated or their use was not advisable, a topical calcineurin inhibitor (pimecrolimus 1% cream or tacrolimus 0.1% or 0.03% ointment) was also allowed. Rescue medication could be used for a maximum of 14 days, and the product, dose, frequency, and duration of therapy were recorded. Patients who received systemic corticosteroids or non-steroidal systemic immunosuppressive or biologic drugs for AD had to discontinue study treatment.
PatientsPatients eligible for the study were adults (age 18–65 years) with a diagnosis of AD for at least 1 year at the screening visit. Patients were required to have moderate-to-severe AD at randomization, defined as Eczema Area and Severity Index (EASI) score ≥ 16, affected body surface area (BSA) ≥ 10%, vIGA-AD score ≥ 3, and average Peak Pruritus NRS ≥ 4 in the 7 days before randomization. Patients had to have a documented history of inadequate response to treatment with TCS or were unsuitable for TCS treatment (e.g., because of important side effects or safety or tolerability considerations).
Patients were excluded if they had received: topical or systemic antihistamines or topical treatments for AD within 7 days before randomization, systemic immunosuppressive or immunomodulating therapy within 4 weeks before randomization, or biologic drugs within five half-lives or 12 weeks before randomization, whichever was longer. Full inclusion and exclusion criteria are provided in Supplementary Tables S2 and S3.
AssessmentsThe primary efficacy endpoint was a composite of EASI-75, no discontinuation of study medication for reasons related to lack of efficacy, no rescue medication use during the 4 weeks before Day 84, and no use of systemic AD treatment. Secondary endpoints included: vIGA-AD response at Week 12, defined as vIGA 0 or 1 with an improvement of ≥ 2 points from baseline; weekly average Peak Pruritus NRS response at Week 12, defined as an improvement (reduction) from baseline of ≥ 4 points in the weekly average Peak Pruritus NRS score, recorded daily by the patient on a handheld device; percentage change in EASI score and weekly average Peak Pruritus NRS percentage change from baseline at Week 12; and absolute change in BSA affected by AD. Blood samples for pharmacokinetic assessment of zabedosertib and its pharmacologically inactive metabolite (BAY 2822815) were taken at each study visit during the treatment phase and at end of treatment.
Safety assessments included recording of adverse events (AEs), clinical laboratory values, vital signs, and electrocardiogram parameters. Confirmed or suspected severe invasive bacterial infections, systemic hypersensitivity reactions, non-invasive infections of the skin, and unexplained cases of rhabdomyolysis were prespecified as AEs of special interest (AESIs) based on the known mode of action of zabedosertib and findings from other IRAK4 inhibitors under development [16].
Sample Size Calculation and Statistical AnalysisAssuming an EASI-75 responder rate in zabedosertib-treated patients of 49% and in placebo-treated patients of 16%, evaluable data from 57 patients (zabedosertib, n = 38; placebo, n = 19) would provide ≥ 90% power to establish superiority of zabedosertib. Based on these assumptions, planned enrollment was 72 patients to obtain 57 evaluable patients overall.
Patients were analyzed according to the treatment they actually received. The main efficacy analysis was performed on the per-protocol set (PPS). These analyses assessed efficacy based on ≥ 80% compliance with study intervention and ≥ 8 weeks length of the treatment phase. Use of rescue medication in the PPS was considered an intercurrent event indicating lack of efficacy after Day 56 to account for a potential late onset of treatment effect. For supplementary analyses on the full analysis set (FAS), use of rescue medication was considered an intercurrent event starting from Day 1 onwards; these analyses were also more conservative regarding the effect size (see Supplementary Statistical Methods section for the exact definitions of PPS and FAS).
The main estimand for the primary and secondary responder endpoints (PPS) was the estimated difference in the proportion of responders between 120 mg zabedosertib and placebo. Use of topical rescue medication from Day 56 onwards, use of systemic standard of care for AD, and discontinuation of treatment due to lack of efficacy were handled as non-responses (composite strategy). Please refer to Supplementary Table S1 for handling of further intercurrent events. Bayesian inference was used to quantify the difference in responder frequencies.
The main analysis for the continuous secondary efficacy endpoints was performed on the PPS using the same strategies to address intercurrent events as described for the primary and secondary binary endpoints above. The mean difference between the treatment arms was estimated using analysis of covariance models. Please see Supplementary Statistical Methods for details.
Safety endpoints were analyzed in the safety analysis set (SAS), which included all patients who received at least one dose of zabedosertib or placebo.
Ethical ApprovalThe study was performed in accordance with the Helsinki Declaration of 1964 and its later amendments. All participating study sites received approval from an institutional review board (Germany: AM-2022-017, Ethik-Kommission bei der Landesärztekammer Baden-Württemberg, Liebknechtstraße 33, 70565 Stuttgart, Germany). All subjects provided informed consent to participate in the study.
Comments (0)