
Remember me

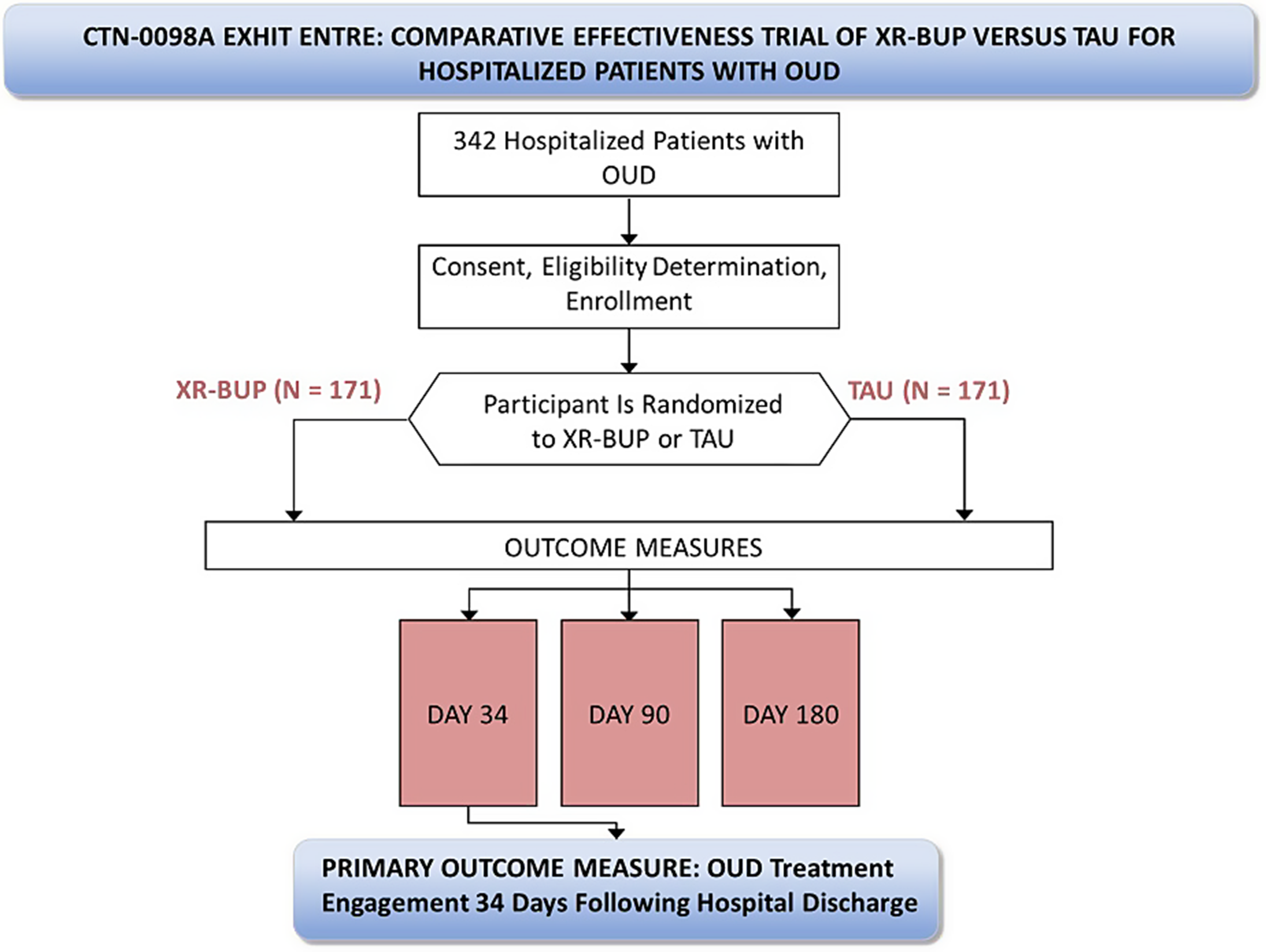

Funded by the NIH HEAL Initiative® and conducted through a cooperative agreement in the National Institute on Drug Abuse (NIDA) National Drug Abuse Treatment Clinical Trials Network (CTN), CTN-0098A Exemplar Hospital Initiation Trial to Enhance Treatment Engagement (EXHIT ENTRE) is a multi-site open-label randomized comparative effectiveness trial of XR-BUP versus TAU for hospitalized patients with untreated OUD prior to hospital admission who are willing to initiate MOUD while hospitalized. The primary objective of this study compares MOUD engagement 34-days following hospital discharge in hospitalized patients randomized to ACS TAU versus a single injection of a 28-day formulation of XR-BUP prior to discharge. Secondary objectives compare safety, engagement in MOUD care at 90- and 180-days post discharge, drug use, hospital readmissions, and emergency room visits among participants randomized to TAU versus XR-BUP.
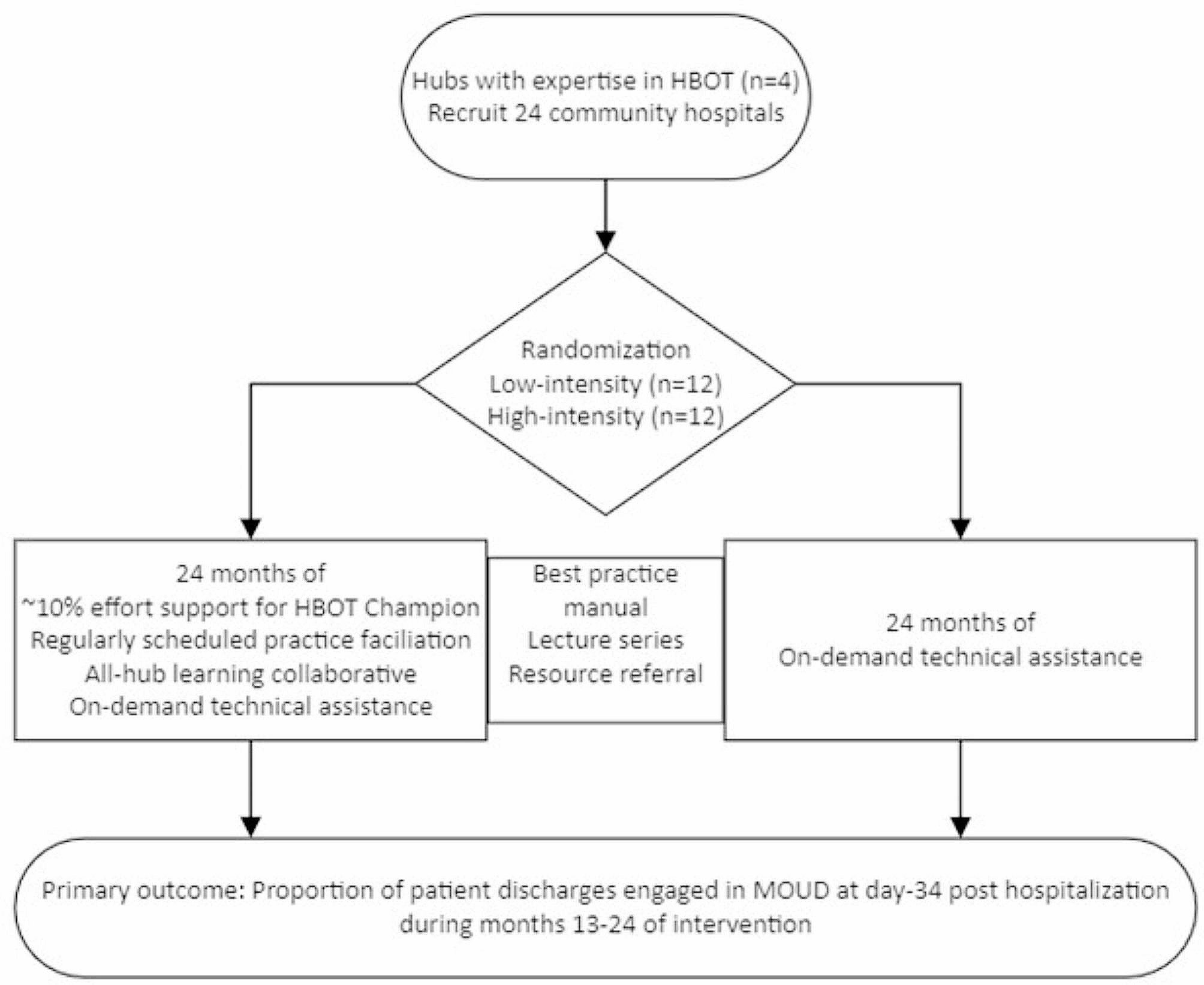
Advarra, a single independent commercial institutional review board (IRB), approved the study (Advarra Pro00047336), with all sites ceding to this single IRB. The study was also reviewed by an independent Protocol Review Board and subsequently a Data and Safety Monitoring Board (DSMB) appointed by the NIDA Center for the Clinical Trials Network (NIDA CCTN). The sponsor does not make decisions regarding publication of this or other study-related manuscripts (Fig. 1).
Fig. 1 Study setting and site selection
Study setting and site selectionSolicitation for hospitals to participate in the study are distributed through the NIDA CTN. To be eligible for participation, hospitals must have established ACS with the capacity to initiate all FDA-approved MOUD. Five hospitals with established ACS were recruited initially, with a sixth hospital added after study initiation to improve recruitment (for sites see https://clinicaltrials.gov/study/NCT04345718).
Participating sites must:
1.Have an existing ACS with experience initiating MOUD prior to hospital discharge.
2.Have a buprenorphine waivered provider “champion” on the ACS who can commit the time required to oversee medical aspects of the study, perform medical assessments, confirm participant eligibility, order study medications, and evaluate and respond to adverse events that may occur during the study period (study protocol was developed prior to the 2022 X-waiver elimination).
3.Have at least one other buprenorphine waivered provider able to perform medical aspects of this study.
4.Have the staff required to recruit and retain participants.
5.Have the staff required to collect research data (including biological specimens such as blood and urine).
6.Have access to local laboratory services to process screening and monitoring specimens (e.g., blood and urine).
7.Be able to manage (store, dispense, and dispose) study medication including having an XR-BUP Risk Evaluation and Mitigation Strategy (REMS) participating pharmacy, if required.
Study participantsEligible participants must be:
1.Hospitalized.
2.At least 18 years of age.
a.Children are not included because addiction health services for them present different challenges and their access to methadone treatment is greatly restricted thus preventing adequate outcomes analysis.
3.Meet DSM-5 criteria for moderate or severe OUD.
4.Willing to initiate MOUD, including buprenorphine.
5.Able to speak English sufficiently to understand the study procedures and provide written informed consent to participate in the study.
Participants are excluded from participation if:
1.Anticipated hospital length of stay less than 24-hours as determined by the ACS.
2.Affected by a serious medical, psychiatric, or substance use disorder that, in the opinion of the study physician, would make it unsafe to participate in the study or may prevent collection of study data. This may include:
a.Disabling terminal diagnosis for which discharge from hospital is not anticipated or for which hospice care is being sought.
b.Severe alcohol or benzodiazepine use disorder that is anticipated to require complex medical detoxification which cannot be completed prior to randomization.
3.Taking a long-acting opioid other than buprenorphine (e.g., methadone, extended-release oxycodone, extended-release morphine) for each of the three days immediately preceding randomization.
4.Liver enzyme tests (Aspartate Aminotransferase (AST), Alanine Aminotransferase (ALT)) more than 3 times upper limit of normal.
5.Currently pregnant or breastfeeding.
6.Known allergy to buprenorphine or components of FluidCrystal delivery system.
7.Receipt of MOUD in the 14 days prior to hospitalization as maintenance treatment; however, patients may have received MOUD for withdrawal management during or prior to hospitalization at the time of enrollment.
8.Are currently in jail, prison or other overnight facility as required by court of law and/or is considered a prisoner under local law or is under current terms of civil commitment or guardianship.
9.Previously randomized as a participant in the study – individuals may only be enrolled and randomized once.
Participant recruitment and consentRecruitment procedures vary by site, but potential participants are drawn from each hospital’s pool of inpatients who have been identified by the ACS as having a possible OUD. Clinical or research staff approach these patients during their index admission to provide information about the study. Individuals providing verbal consent are prescreened under a HIPAA Waiver of Authorization by reviewing the electronic medical record to further assess inclusion/exclusion criteria. If a person meets initial eligibility criteria during the prescreening process, they give written informed consent prior to completing screening and baseline procedures. The informed consent process includes a quiz testing comprehension of study activities, alternatives, and potential risks and benefits, which may be repeated until answered correctly. Participants can opt to provide additional consent for blood collection and biobanking for potential future addiction- and MOUD- related genetic studies. Baseline data collection is completed prior to randomization.
RandomizationApproximately 342 eligible participants are randomized in a 1:1 ratio to open-label XR-BUP or TAU (e.g., methadone, sublingual buprenorphine, extended-release naltrexone). The randomization process is performed by computer by a central data and statistics center (DSC). A permuted block randomization procedure with random block sizes is used. The DSC statistician generates the randomization schedule using blocks of varying sizes within strata (hospitals) to ensure lack of predictability along with relative equality of assignment across treatment groups. The randomization details such as block size are not conveyed to staff or participants. The DSC statistician reviews randomization data on a regular basis to ensure that the scheme is being implemented according to plan. A randomization slot, once used, is not re-allocated.
InterventionThis study randomly assigns participants to ACS TAU or a single injection of 28-day formulation XR-BUP administered within 72 h of anticipated hospital discharge. This study is open-label and not blinded. There are no study interventions that extend beyond the index hospitalization or to facilitate care post-hospitalization or MOUD adherence beyond the standard care provided by the ACS. The ACS may initiate MOUD at any point during hospitalization. TAU may consist of any FDA-approved MOUD (other than XR-BUP) with the dose and amount of MOUD supplied upon hospital discharge determined by the ACS. For methadone, federal regulation prevents more than three days of dispensing outside of an opioid treatment program, whereas up to 30-days of buprenorphine may be provided upon hospital release. Although participants randomized to XR-BUP do not receive their XR-BUP injection until they are within 72-hours of anticipated hospital discharge, the ACS may offer other MOUD during their hospitalization per local standard of care, which reduces potential imbalance between arms in approaches to MOUD during recruitment and screening. The rationale for not providing XR-BUP until just prior to hospital discharge is to account for variable length of hospital admissions and prevent some participants receiving an injection early in a prolonged hospitalization where the effect of XR-BUP may wear off during hospitalization or shortly thereafter.
MOUD initiation strategies are determined by the ACS and doses titrated per their standard of care. Participants randomized to XR-BUP must tolerate at least one dose of sublingual buprenorphine prior to injection. As XR-BUP is available in three doses, participants can receive a dose commensurate with their current (if on a stable dose) or anticipated stable dose of sublingual buprenorphine. Available doses of XR-BUP (equivalent sublingual dose range) are: 64 mg (8–10 mg), 96 mg (12–16 mg), and 128 mg (18–24 mg). Because the lowest XR-BUP dose approximates 8 mg sublingually, participants are required to have an anticipated maintenance dose of at least 8 mg sublingually. Access to XR-BUP (CAM-2038, Braeburn, Inc.) is via an Investigational New Drug Application as it was not commercially available at time of study initiation, although it has since become available. The National Institute on Drug Abuse negotiated with Braeburn Inc. to procure donated XR-BUP for this study, the lead investigators were not part of this process.
Participant follow-up for outcome assessments occur 34-, 90-, and 180-days following discharge from the index hospitalization. Participants receive $50 for baseline assessment and enrollment and $50, $75, and $100, respectively for the three follow-up visits.
AssessmentsStudy assessment and the schedule of study activities are presented in the Table. Where possible, assessments include validated tools that are used widely throughout other NIDA CTN studies. For example, the Substance Abuse and Addiction Collection of the PhenX Toolkit [30] is used at baseline to assess demographics (age, ethnicity, sex, race, educational attainment, employment status and marital status), BMI, Human Immunodeficiency Virus (HIV) Risk & Status, and substance use measures including age of onset, past 30-day quantity and frequency, and lifetime use for alcohol, tobacco and other substances.
While the Timeline Followback (TLFB) [31, 32] is used at each study visit to assess past month drug and alcohol use, we have adapted an additional TLFB that asks about daily use of MOUD in the specified timeframe for the 34, 90, and 180-day follow-up visits.
Tests for viral hepatitis (hepatitis B surface antigen and antibody, hepatitis C antibody with reflex PCR, if positive) and HIV with reflex viral load, if positive, are collected at baseline unless results from the 90-days prior to admission can be abstracted from the medical record.
Urine drug screens (UDS) are collected at screening/baseline and at each follow-up visit to assess secondary outcomes, unless the follow-up visit is fully remote. All urine specimens are collected using FDA-approved one-step temperature-controlled urine drug test cups testing for the presence of: opiates, oxycodone, barbiturates, benzodiazepines, cocaine, amphetamines, methamphetamines, marijuana, methadone, buprenorphine, phencyclidine (PCP), fentanyl, and ecstasy (MDMA). Results of UDS are not shared with participant clinical care givers.
Medical comorbidity is abstracted from the medical record problem list and/or discharge diagnoses as is the Comorbidity Severity Index (i.e., CMS-HCC Risk Adjustment [33]).
Pre- and post- XR-BUP injection evaluation for precipitated opioid withdrawal uses the Clinical Opioid Withdrawal Scale (COWS) [34] with a 5-point increase indicating precipitated withdrawal. An independent data and safety monitoring board (DSMB) convened by NIDA will have access to safety reports and will meet at least annually. The DSMB may receive aggregate data (blinded) as well as by randomization group (unblinded). The DSMB may recommend protocol modifications or even early study termination if either study arm has a clinically important excess of serious adverse events.
The majority of collected data are directly entered without personal identifiers into a secure electronic case report form (eCRF) system maintained by the DSC (Advantage eClinical). Data abstracted from the electronic health record or requiring a paper source CRF are transcribed from paper CRF to Advantage eClinical by study staff. Following study completion and publication of the primary outcome paper, as per NIH and HEAL Initiative policy, study data will be available through the NIDA data repository (Table 1).
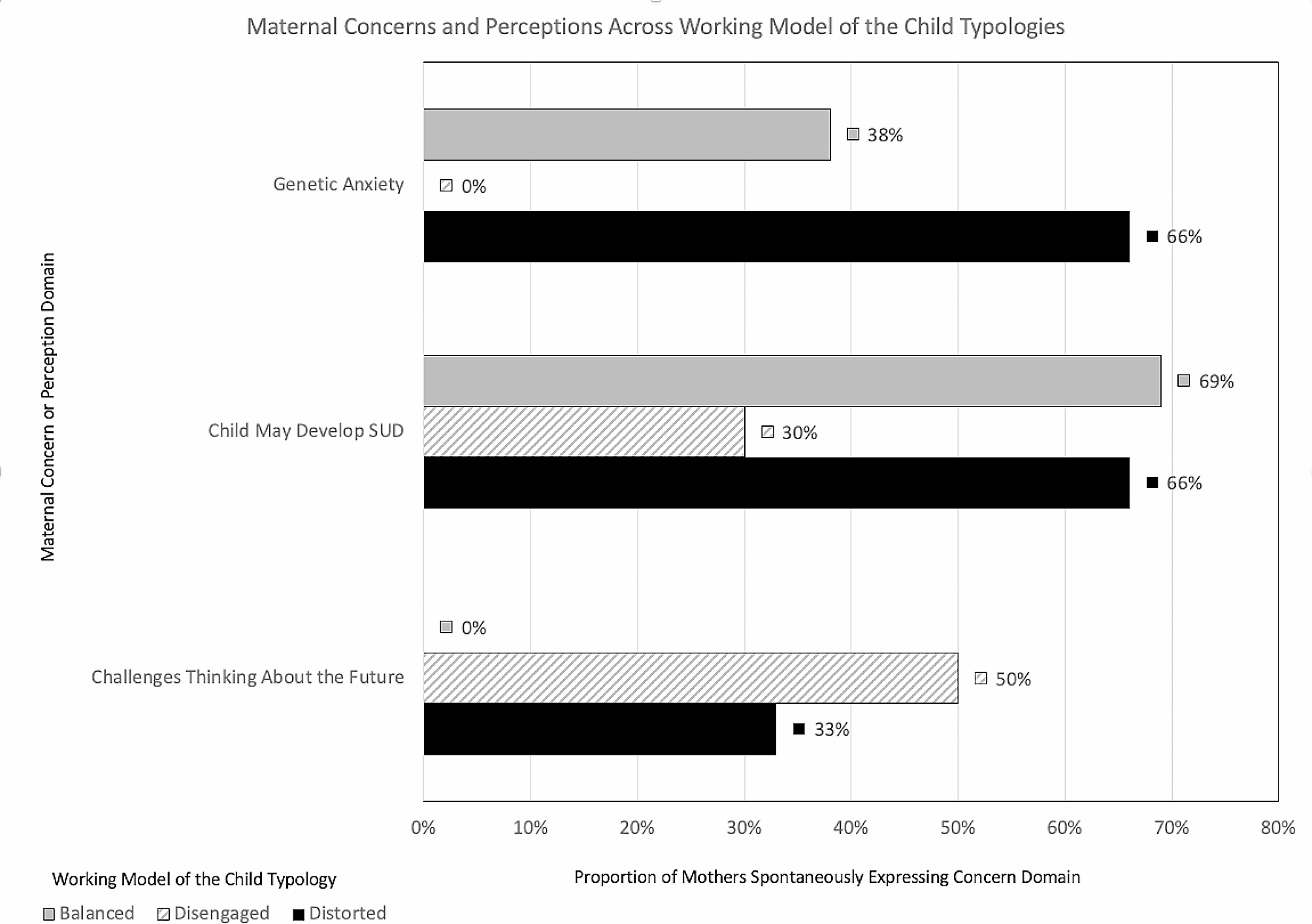
Table 1 Table of assessmentsOutcomesThe primary outcome measure is the proportion of participants engaged in MOUD care on the 34th day following hospital discharge. Engagement in MOUD care is defined as verifiable coverage with a prescribed MOUD on that 34th day regardless of the source of prescribed MOUD coverage (e.g., opioid treatment program, primary care, jail, etc.). Participants give appropriate permission for release of such data during informed consent procedures and/or during study follow-up visits. Other objective means of MOUD verification, such as bringing a prescription bottle with participant name and dates covered by the prescription can, also be used for primary outcome.
Data for primary outcomeMOUD engagement on the 34th day post hospital discharge is collected by self-report verified with an objective data source (e.g., pill bottle, prescription record, provider confirmation upon release of information, etc.) or, in the absence of self-report, an objective data source is used for the primary outcome (e.g., electronic medical records, provider confirmation upon release of information, etc.).
Secondary outcomes include:
1.Proportion of participants that experience any treatment emergent AEs and SAEs (those developed after initiation of treatment) following hospital discharge.
2.Proportion of participants engaged with MOUD 90- and 180-days following hospital discharge.
3.Proportion of participants with positive urine drug test for opiates, fentanyl, and oxycodone 34-, and 90-, and 180-days following hospital discharge.
4.Proportion of participants with self-reported non-prescribed opioid use 34-, and 90-, and 180-days following hospital discharge.
5.Proportion of participants with self-reported hospital readmissions at the 34- and 90-day visits.
6.Proportion of participants with self-reported ED visits at the 34- and 90-day visits.
Exploratory outcomes include:
1.Proportion of participants with positive urine drug test for illicit drugs 34-, and 90-, and 180-days following hospital discharge.
2.Proportion of participants with self-reported non-prescribed drug use 34-, and 90-, and 180-days following hospital discharge.
3.Change in ASI-Lite measures from baseline to 180 days.
4.Proportion of participants with self-reported medical follow-up at the 34- and 90-day visits.
5.Proportion of participants adherent to antibiotic for OUD-related infections (e.g., endocarditis or skin-soft tissue infections) (when applicable).
6.Self-reported alcohol use 34-, and 90-, and 180-days following hospital discharge.
7.Hospital length of stay (for both the index hospitalization and readmissions).
8.Self-reported 180-day hospital readmission rates.
9.Self-reported 180-day ED visit rates.
10.All-cause mortality rates at 30-, 90- and 180-days following hospital discharge.
11.Comparative cost-effectiveness of XR-BUP and other MOUD.
12.Non-fatal opioid overdose rates (discharge to 34-day visit and since last study visit at the 90- and 180-day visits).
13.Fatal opioid overdose rates up to the 180-day visit.
14.Satisfaction with MOUD treatment 180 days following hospital discharge.
15.Self-reported 34- and 90-day hospital readmission rates related to OUD.
16.Self-reported 34- and 90-day ED visits related to OUD.
17.Quality of life.
18.Receipt of subsequent XR-BUP injections (among the XR-BUP group only).
19.Receipt of other MOUD treatments during follow-up (e.g., XR-BUP in the control group, methadone or SL-BUP in the XR-BUP group).
Sample size342 participants will be randomized in a 1:1 ratio to XR-BUP or TAU. The power calculation assumes the 34-day engagement rate is 0.39 for TAU [22] and 0.60 for XR-BUP and that 15% of the primary outcome values will be missing and treated as “not engaged” for the primary analysis. In that scenario, 85% of patients will have the assumed rate (0.39 and 0.60 for TAU and XR-BUP, respectively) and 15% of patients in both groups will have a rate of 0.0. As such, the new engagement rates will be 0.3315 for TAU and 0.51 for XR-BUP.
A Cochran-Mantel-Haenszel test with continuity correction based on odds ratios (OR) of 2.1 assuming different engagement rates by site (6 sites with 34-day engagement rates of 0.29 up to 0.37 for TAU and 0.46 up to 0.55 for XR-BUP group) suggests that a total of 342 participants (171 participants in each group) need to be enrolled to have 90% power to detect a difference in the 34-day engagement rates between TAU and XR-BUP (two-sided test, alpha [false positive rate] = 0.05). Following a blinded interim analysis, the DSMB may recommend the protocol be amended to increase the number of subjects enrolled into the study.
Outcomes analysesThe primary analysis compares the two groups (TAU and XR-BUP) for the primary outcome (engagement in MOUD care on the 34th day following hospital discharge) using the Cochran-Mantel-Haenszel test stratified by hospital under the principle of intention-to-treat (ITT), i.e., participants are analyzed in the group to which they were randomized irrespective of whether they received their assigned treatment. An additional analysis may be performed using a per-protocol analysis population (i.e., according to the actual treatment received).
Supplementary analyses of the primary outcome will be performed as follows: (a) consider gender as an effect modifier, (b) add adjusters for possible differences between groups, and (c) adjust for missing outcome data. Logistic regression is used for (a) and (b) whereas analyses adjusting for missing outcome data will use inverse probability weighting (IPW).
Analyses of secondary outcomes, broadly, will have a form similar to analysis of the primary outcome, or outcomes measured at multiple time-points (e.g., engagement in OUD care at 34-, 90-, and 180-days) will use a mixed-effect analysis with a person-specific random effect to capture correlation of a person’s multiple time-points. The particular form of analysis, e.g., logistic regression vs. normal-errors regression, will depend on the outcome, and outcomes on continuous or effectively continuous scales may be transformed before analysis (e.g., by taking the logarithm) to allow use of analyses that assume normally distributed errors.
Analytic approaches for exploratory outcomes are similar to those described for the primary and secondary outcomes. In addition, we will explore moderators and mediators of MOUD engagement and other outcomes such as hospital length of stay and antibiotic completion rates (where appropriate). Potential moderators and mediators include hospital length of stay, post-hospital discharge medical follow-up, quality of life, depression, post-traumatic stress syndrome, pain, and treatment satisfaction.
Cost-effectiveness analysis will be conducted from a health sector perspective over the 6-month trial participation time horizon as well as over a remaining lifetime time horizon (by extrapolating from trial outcomes). Costs will include the healthcare costs associated with the initial OUD management pre-discharge (either a single dose of XR-BUP or TAU) and post-discharge healthcare utilization (ED visits and hospitalizations) and OUD care in the first six-months. Post-discharge costs will be estimated by multiplying self-reported healthcare utilization by the relevant Centers for Medicare and Medicaid Services (CMS) reimbursement rates [42].
The effectiveness of the interventions will be assessed in terms of quality-adjusted life-years (QALYs), which reflect both length and quality of life. We will estimate the average QALYs accrued under each intervention by calculating the amount of time participants spend in different OUD states (based on reported ED use, hospitalizations, and OUD care in the first 6 months) multiplied by the health-related utility of that state estimated from published data [10, 43]. Mortality is accounted for by assigning death a utility value of zero.
For the lifetime time horizon analysis, costs and QALYs after 6 months will be estimated by extrapolating from trial outcomes using estimated OUD outcomes from the published literature, economic analyses of MOUD treatments, and OUD modeling studies. We will conduct sensitivity analyses to assess the sensitivity of our cost-effectiveness conclusions on any extrapolation assumptions.
Once costs and QALYs have been assessed for XR-BUP and TAU, we will compare the incremental costs and benefits of XR-BUP. If XR-BUP yields a health benefit (increase in QALYs) and is less expensive than TAU, we will calculate the extent of this cost-savings to the healthcare sector. If XR-BUP is more expensive, but also more beneficial, than TAU, we will calculate the incremental cost-effectiveness ratio (ICER) of XR-BUP, which is the additional cost of each additional QALY gained by using XR-BUP for hospital-initiated MOUD instead of TAU. Cost-effectiveness conclusions will depend on the value of the ICER. In the US, interventions with an ICER less than $100,000 per QALY gained are considered cost-effective, between $100,000 - $150,000 per QALY gained are considered marginally cost-effective, and over $150,000 per QALY gained is considered not cost-effective.
Trial progressThe study was approved by a single IRB in 2021 and recruitment is ongoing with more than 265 participants recruited by July 2024. Recruitment in hospitals was challenging during peak COVID-19 emergencies and supply chain disruptions led to a 6-week interruption in access to XR-BUP in 2022. The protocol is in its fifth version, with minor corrections to the previous versions.
Comments (0)