
Remember me




A male child was born at 36 + 4 weeks gestational age by spontaneous vaginal delivery complicated by shoulder dystocia with APGARs of 6/8/8 at 1/5/10 minutes of life, respectively. The mother had gestational diabetes and he was large for gestational age with a weight of 4.17 kg. Shortly after birth, he developed respiratory failure, hypotensive shock and rapidly declining hematocrit (<15%) with concerns for disseminated intravascular coagulation. His white blood cell count (WBC) peaked at 27.5 × 103/mL before declining to 1.5 × 103/mL, and his platelets nadired at 23 × 103/mL. Blood cultures were negative. An abdominal ultrasound identified a left renal vein thrombosis and a large left adrenal hemorrhage measuring 5.3 × 5.2 × 3.6 cm, the likely etiology of his decompensation. He required multiple blood product transfusions for resuscitation and completed a 7-day course of antibiotics for culture-negative sepsis, with clinical stabilization. A repeat abdominal ultrasound on day of life 14 demonstrated resolution of the renal vein thrombosis and a decrease in size of the adrenal hematoma. He was hospitalized for 6 weeks in the neonatal intensive care unit and his clinical course was complicated by medical necrotizing enterocolitis on day of life 10, cholestatic hepatitis, mild hypoxic ischemic encephalopathy and poor weight gain despite placement of a gastrostomy tube and fortification of enteral feeds.
On the day of discharge at 6 weeks of life, he had a routine follow-up abdominal ultrasound to evaluate the interval evolution of his adrenal hematoma, which unexpectedly revealed a large cystic mass measuring 9.0 × 5.5 × 6.6 cm that occupied much of the left hemiabdomen with displacement of the left kidney across the midline. There was a new concern for malignancy, especially cystic neuroblastoma, but the parents refused further inpatient evaluation and insisted on hospital discharge. He was evaluated by oncology 1 week after discharge, but additional evaluation for neuroblastoma, including urinary homovanillic and vanillylmandelic acid levels and abdominal computed tomography imaging, was unsuccessful secondary to social and technical issues that arose during care. His gastrostomy tube was removed at parental insistence because of persistent leakage with each feed. He had no fevers, respiratory distress or systemic symptoms reported by the family, and his feeding intolerance was consistent with mass effect from the adrenal lesion.
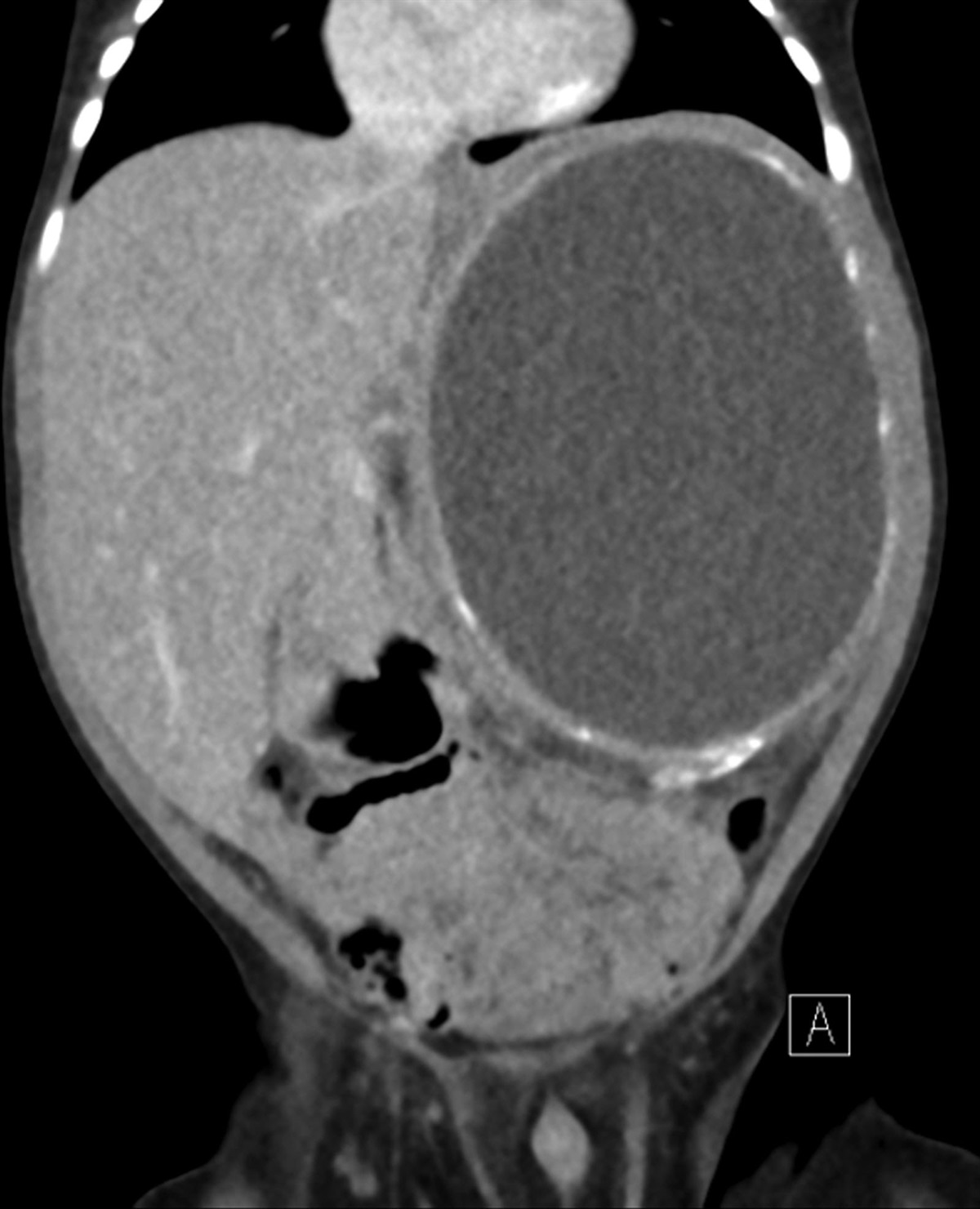
He was readmitted to our institution at 2.5 months of age for his failure to thrive and progressive abdominal distension. At the time of admission, his temperature was 36.5 °C, heart rate 130 beats per minute, respiratory rate 30 per minute and pulse oximetry 99% on room air. His exam was notable for jaundice and marked abdominal distension with formula leakage from a gastrocutaneous fistula at the site of his former gastrostomy tube. Crepitus was noted on palpation of the abdomen, which was suspected to be related to the gastrocutaneous fistula. The remainder of his physical exam was normal, and he was behaving per his baseline according to the parents. An abdominal computed tomography scan redemonstrated a large retroperitoneal cystic mass with peripheral calcifications (Fig. 1). Laboratory findings revealed a total bilirubin of 8.6 mg/dL, direct bilirubin 4.4 mg/dL, alanine aminotransferase 196 U/mL and aspartate aminotransferase 221 U/mL, each of which was downtrending from his prior hospitalization. His WBC count was 22.9 × 103/mL, hemoglobin 7.7 g/dL, platelets 605 × 103/mL and urinary homovanillic and vanillylmandelic acid levels were normal. A nasojejunal tube was placed for feeding.
 FIGURE 1.:
FIGURE 1.: Abdominal computed tomography scan. A large, centrally hypoattenuating, peripherally calcified mass measuring 8.0 × 7.6 × 10.1 cm is identified in the left hemiabdomen. The left kidney is not visualized.
On the fourth hospital day, the patient became febrile and there was concern for sepsis. The WBC was stable from admission at 23.3 × 103/mL (49% neutrophils, 11% bands, 29% lymphocytes, 8% monocytes and 4% eosinophils), but his blood culture was positive for Escherichia coli. The isolate was resistant to cefazolin, and in view of his cholestasis, he was started on intravenous cefepime. His urine culture was negative. On the ninth hospital day, interventional radiology placed an 8-French Dawson-Mueller catheter into the cystic mass with drainage of 330 mL of purulent fluid. A culture of this fluid was also positive for E. coli.
He completed a 14-day course of intravenous cefepime for his bacteremia. A repeat abdominal ultrasound revealed a heterogeneous mass measuring 4.9 × 5.8 × 4.2 cm, with no obvious persistent fluid collection. He was transitioned to oral amoxicillin/clavulanate (90 mg/kg/d of amoxicillin component) at discharge with the retroperitoneal drain still in place.
In the outpatient setting, he had no further fevers and his weight gain slowly improved. One month after discharge, he had a follow-up ultrasound that demonstrated a persistent mass measuring 5.6 × 3.2 × 4.1 cm. He was having minimal output from his drain, but his WBC remained elevated at 22.6 × 103/mL. His antibiotic regimen was changed to cefdinir and metronidazole and 3 weeks later he underwent surgical excision.
A 6.0 × 5.0 × 3.0 cm tan nodular mass with 20% gross necrosis was excised from the retroperitoneum. There was significant local inflammation with dense adhesions to the diaphragm requiring an en bloc partial resection of the medial and posterior diaphragm. No identifiable kidney structure was evident. Histologic analysis revealed the diagnosis.
DENOUEMENTCytologic staining demonstrated histiocytes with rounded cytoplasmic inclusions. Histopathology demonstrated intra- and extracellular targetoid inclusions, which were highlighted with von Kossa stain, a calcium stain (Fig. 2). These findings were consistent with Michaelis-Gutmann bodies and pathognomonic for a diagnosis of malakoplakia. Fungal and acid-fast bacilli stains and cultures were negative. He demonstrated marked clinical improvement after resection, and antibiotics were discontinued 2 weeks later when a follow-up abdominal ultrasound demonstrated complete resolution of the mass.
 FIGURE 2.:
FIGURE 2.: Histopathologic evaluation. Von Kossa stain at ×20 magnification. The stain (black) highlights the Michaelis-Gutmann bodies (rounded calcified phagosomes).
Malakoplakia is a very rare granulomatous disease that most commonly affects immunocompromised individuals.1 The pathogenesis is incompletely understood and likely multifactorial, including the infecting organism, underlying host immune status and abnormal macrophage response to infection with impaired lysosomal function.2 The failure to effectively kill microorganisms leads to calcium and iron deposition on bacterial fragments and accumulation of phospholipids and microvesicles resulting in the characteristic lamellar Michaelis-Gutmann bodies.2–4 Phagocytosis does not appear to be impacted. Prior reports have demonstrated low intracellular cyclic-guanosine monophosphate in patients with malakoplakia who were able to control the infection following administration of bethanechol chloride, a cholinergic agonist,5,6 though this has not been consistently demonstrated.7
Malakoplakia was first described in humans by von Hansemann in 1901 followed by collaborators Michaelis and Gutmann in 1902.8 The first 4 patients had bladder involvement with plaque-like lesions, which led von Hansemann to coin the name malakoplakia from the Greek words malakos (soft) and plakos (plaque).8 The genitourinary tract is the most common location of malakoplakia in adults, with a female preponderance, though involvement of most organ systems has been reported. E. coli is the most implicated organism.2 Reports in pediatric patients are limited and many providers may not be familiar with this diagnosis. This is the second case of adrenal malakoplakia in a child. The first case also occurred in an infant with a history of adrenal hemorrhage.9
Malakoplakia may mimic other conditions, including inflammatory bowel disease, familial polyposis coli or malignancy,10 and pathology is necessary to establish the diagnosis. Important differential diagnoses of retroperitoneal masses include megalocytic interstitial nephritis, renal giant cytoplasmic inclusion (as seen in Chediak-Higashi syndrome), xanthogranulomatous pyelonephritis, mycobacterium infection or Langerhans cell histiocytosis.2,11 When malakoplakia involves the gastrointestinal tract, cross-reactivity with Trophyerma whipplei immunostaining may occur.11 In our patient, the unexpected evolution of his adrenal hemorrhage was concerning for an underlying malignancy, especially neuroblastoma, but evaluation was negative.
Malakoplakia may cause significant morbidity and mortality,12 and treatment consists of prolonged antibiotic therapy and potentially surgical resection, especially for larger lesions and when there is compression of vital structures.2 Antimicrobial therapy should be directed at the infecting organism (if known) with agents that concentrate intracellularly, such as trimethoprim-sulfamethoxazole, fluoroquinolones or rifampin.2,5,8,11 Prior studies on the management of malakoplakia have identified improved cure rates with fluoroquinolones13 and improved response to treatment after switching to agents that concentrate intracellularly.14,15
In patients on immunosuppressive therapy, reduction of immunosuppression, if possible, should be considered. Biggar et al demonstrated improvement in 4 patients with malakoplakia secondary to E. coli after tapering or discontinuing azathioprine and prednisone. In all 4 patients, malakoplakia persisted despite long-term antimicrobial therapy and cholinergic agonists, but improved with the reduction of immunosuppression. Each of the patients had abnormal monocyte and neutrophil bactericidal activity that normalized after tapering or discontinuing immunosuppressive therapy.7
In the present case, it is suspected that the child’s adrenal hemorrhage became secondarily infected during the episode of necrotizing enterocolitis with subsequent development of malakoplakia and chronic progressive infection. His critical illness and young age may have contributed to a dysregulated immune response, complicated by poor weight gain, leading to malakoplakia. Drainage of the abscess improved the compressive symptoms of the mass, but there was no further reduction in the solid component of the lesion despite 10 weeks of antibiotic therapy. It is possible that a better response to therapy would have been seen if an antibiotic with improved intracellular concentration was used, but based on the size of his lesion surgical resection was the preferred approach. After surgical resection, he had excellent catch-up weight gain, normal neurodevelopment and resolution of his cholestatic hepatitis. He required surgical intervention for a diaphragmatic hernia repair 18 months after his initial surgery, but he has had no further serious infections.
In conclusion, malakoplakia is a rare granulomatous disorder, but it is an important consideration in the differential diagnosis of abdominal masses not responding as expected to targeted therapy. It may be indolent or rapidly progressive, may mimic neoplasm and is often associated with Gram-negative bacilli infections, especially E. coli. Our case highlights the challenges of diagnosis and appropriate therapy without histologic evaluation.
REFERENCES 1. Archer SR, Abramowsky CR, Kobrynski L, et al. Malakoplakia and primary immunodeficiency. J Pediatr. 2014;165:1053–1056. 2. Yousef GM, Naghibi B, Hamodat MM. Malakoplakia outside the urinary tract. Arch Pathol Lab Med. 2007;131:297–300. 3. Lou TY, Teplitz C. Malakoplakia: pathogenesis and ultrastructural morphogenesis: a problem of altered macrophage (phagolysosomal) response. Hum Pathol. 1974;5:191–207. 4. van Crevel R, Curfs J, van der Ven AJ, et al. Functional and morphological monocyte abnormalities in a patient with malakoplakia. Am J Med. 1998;105:74–77. 5. Abdou NI, NaPombejara C, Sagawa A, et al. Malakoplakia: evidence for monocyte lysosomal abnormality correctable by cholinergic agonist in vitro and in vivo. N Engl J Med. 1977;297:1413–1419. 6. Webb M, Pincott JR, Marshall WC, et al. Hypogammaglobulinaemia and malakoplakia: response to bethanechol. Eur J Pediatr. 1986;145:297–302. 7. Biggar WD, Crawford L, Cardella C, et al. Malakoplakia and immunosuppressive therapy. Reversal of clinical and leukocyte abnormalities after withdrawal of prednisone and azathioprine. Am J Pathol. 1985;119:5–11. 8. Dasgupta P, Womack C, Turner A, et al. Malacoplakia: von Hansemann’s disease. BJU Int. 1999;84:464–469. 9. Sinclair-Smith C, Khan L, Cywes S. Proceedings: malacoplakia in childhood. Arch Dis Child. 1974;49:498. 10. Parkin CJ, Acland G, Sulaiman B, et al. Malakoplakia, a malignant mimic. Bladder. 2020;7:e44. 11. Stanton MJ, Maxted W. Malacoplakia: a study of the literature and current concepts of pathogenesis, diagnosis and treatment. J Urol. 1981;125:139–146. 12. Deridder PA, Koff SA, Gikas PW, et al. Renal malacoplakia. J Urol. 1977;117:428–432. 13. van der Voort HJ, ten Velden JA, Wassenaar RP, et al. Malacoplakia. Two case reports and a comparison of treatment modalities based on a literature review. Arch Intern Med. 1996;156:577–583. 14. Ho KL, Rassekh ZS, Nam SH. Bilateral renal malakoplakia. Urology. 1979;13:321–323. 15. Maderazo EG, Berlin BB, Morhardt C. Treatment of malakoplakia with trimethoprim-sulfamethoxazole. Urology. 1979;13:70–73.
Comments (0)